1,3-Dipolar Cycloadditions
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Planning Organic Syntheses
The essential features of the Diels–Alder reaction are a four-electron π system and a two-electron π system which interact by a HOMO–LUMO interaction.
1,3-DIPOLAR CYCLOADDITIONS
The
essential features of the Diels–Alder reaction are a four-electron π system and a two-electron π system which interact by a HOMO–LUMO
interaction. The Diels–Alder reaction uses a conjugated diene as the
four-electron π system and a π bond between two elements as the
two-electron component. However, other four-electron π systems could potentially interact with olefins in a similar
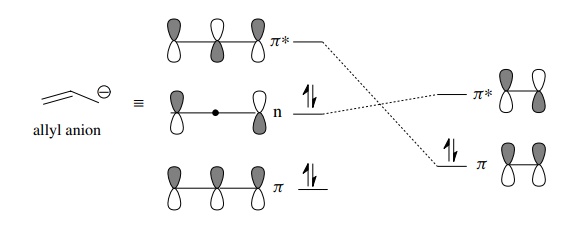
fashion to give cycloaddition products. For example, an allyl anion is a
four-electron π system whose orbital
diagram is shown below. The symmetry of the allyl anion nonbonding HOMO matches
that of the olefin LUMO (as does the olefin HOMO and the allyl anion LUMO);
thus effective overlap is possible and cycloaddition is allowed. The HOMO–LUMO
energy gap determines the rate of reaction, which happens to be relatively slow
in this case.
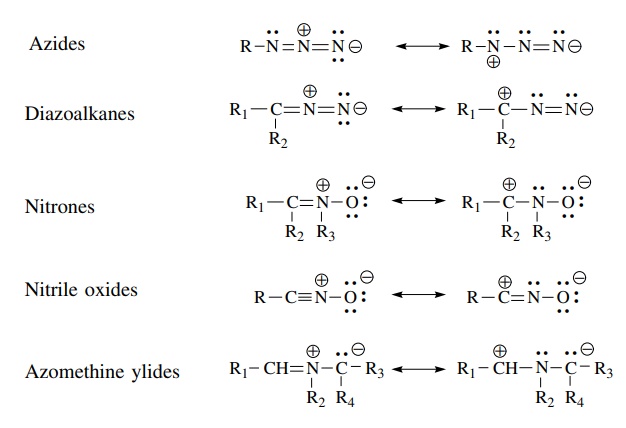
Molecules
isoelectronic with the allyl anion but which are neutral and have at least one
resonance form with formal positive and negative changes in a 1,3 relationship
are called 1,3 dipoles.
All
have an orbital diagram analogous to the allyl anion in which three
inter-acting p orbitals give rise to three molecular orbitals containing a
total of four π electrons. For
example, a nitrone is seen to have a C–N π
bond interacting with a filled orbital on the oxygen atom to define a new π system containing four π electrons.
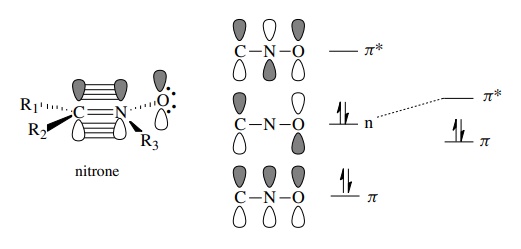
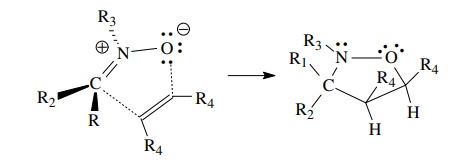
Interaction
of the HOMO of the 1,3 dipole with the LUMO of a simple π bond (called a dipolarophile in this process) leads to bond
formation between the ends of the 1,3 dipole and the olefin, producing a new
five-membered ring.
The
process is a concerted 4π + 2π cycloaddition and is related electroni-cally to the
Diels–Alder reaction. The formal charges are destroyed during the cyclization
and a wide variety of heteroatom components are possible in the 1,3 dipole.
Moreover other π bonds besides
alkenes and alkynes can be used as dipolarophiles. As a result, 1,3 dipolar
cycloadditions have been used to make a large number of heterocyclic compounds.
Since
the 1,3 dipolar cycloaddition is concerted, the reaction is stereospecific and
the geometry of the olefin is maintained in the cyclic product.
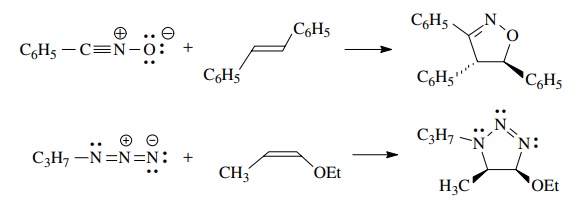
If
a symmetric dipolarophile is used, then no regioisomers are possible. If, however,
the dipolarophile is unsymmetric, then regioisomers are possible.
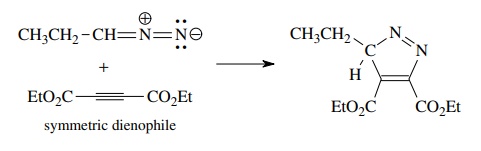
As
in the case of the Diels–Alder reaction, the regioselectivity can be understood
in terms of the electron distribution in the 1,3 dipole and the dipolarophile.
For example, a nitrile oxide should have a relatively electron deficient carbon
and a relatively electron rich oxygen. Reaction with propene, which has
greatest elec-tron density at C-2 because of the inductive effect of the methyl
group, gives the regioisomer C.
Matching the polarity of the dipole and the dipolarophile predicts this
product. Conversely reaction with methyl acrylate, which because of
conju-gation has electron deficiency at C-3, gives regioisomer D as the major product.
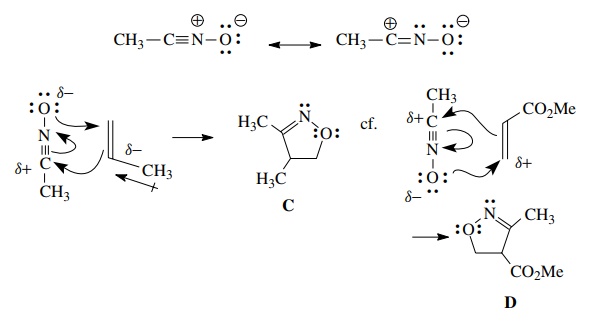
Polarity
matching to predict the major product of 1,3 dipolar cycloadditions is
qualitative only and frequently fails to predict the major product correctly.
This is because each 1,3 dipole tends to exhibit a characteristic
regioselectivity toward particular dipolarophiles that may be modified by
steric and/or strain effects. In fact there is still some uncertainty as to
just what factors do influence the regioselectivity in these systems.
Nevertheless
1,3 dipolar cycloadditions are an important method for the syn-thesis of a wide
variety of heterocyclic compounds. Furthermore they illustrate the generality
of 4 + 2 cycloaddition
reactions as a means to prepare cyclic products efficiently from acyclic
precursors.
To
use 1,3 dipolar cycloadditions in a retrosynthetic sense, it is necessary to
know what 1,3 dipoles are available. The list on pages 319–320 is
repre-sentative of the more common and useful examples, although many others
have been reported. Azides, diazo compounds, and nitrones are normally isolable
com-pounds which can be added to a solution of an olefin. Other 1,3 dipolar
species such as nitrile oxides and azomethine ylides are not stable molecules;
they must be generated in the reaction mixture in the presence of the olefin.
As might be expected, many different ways to generate 1,3 dipoles have been
developed.
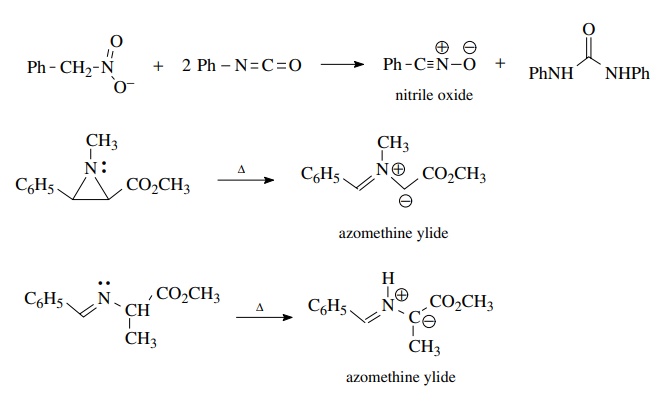
Nitrile
oxides are often generated by the dehydration of nitro compounds by reagents
such as phenyl isocyanate. Azomethine ylides can be generated by the pyrolysis
of aziridines or by the prototopic isomerization of imines upon heating.
The
next step is to identify the five-membered ring which could be assembled by a
1,3 dipolar cycloaddition and then identify the π system and 1,3 dipole needed to give the proper array of
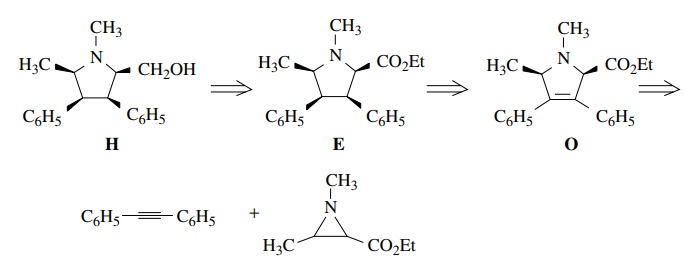
heteroatoms. Thus if the pyrrolidine H
is needed, it is clear the alcohol could be made by reducing the ester function
of E. Also important is the issue of
the all-cis stereochemistry. One way to ensure the all-cis stereochemistry is to do a catalytic hydrogenation of
the olefin O. The delivery of
hydrogen would come from the less hindered face of O and would give the all-cis product. The needed olefin O could be made by a 1,3 dipolar
addition between an azomethine ylide and diphenyl acetylene.
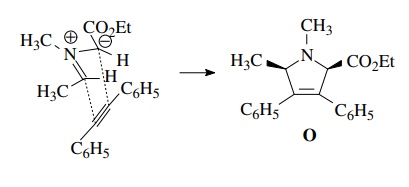
The
only concern is the cis stereochemistry of the cycloadduct O. If the planar azomethine ylide adopts the least sterically
hindered “W” geometry, then the cis isomer will be produced as a pair of
enantiomers. The use of cis-stilbene
as the dipolarophile to obtain the all-cis geometry in one step would require
that only the endo transition state produces product. Although endo transitions
are favored in 1,3 dipolar cycloadditions, mixtures of diastereomers from the
exo and endo transition states are usually formed. Catalytic hydrogenation has
a higher facial selectivity and is much more likely to give a single
diastereomer.
These
are but a few examples of how retrosynthetic analysis can be used to develop
one or more synthetic routes to a target. Developing synthetic strategies is
one of the most creative activities that organic chemists perform. It requires
that many different inputs and conditions be cohesively merged into a single
thematic development that contains elements of texture and beauty, proportion
and balance, and risk and reward. The process is every bit as creative as
painting, sculpting, or writing the great American novel!
Related Topics
