Antiarrhythmic Drugs
| Home | | Pharmacology |Chapter: Essential pharmacology : Antiarrhythmic Drugs
These are drugs used to prevent or treat irregularities of cardiac rhythm.
ANTIARRHYTHMIC
DRUGS
These are drugs used
to prevent or treat irregularities of cardiac rhythm.
Nearly 3 out of 4 patients of acute myocardial infarction (MI)
and about half of those given a general anaesthetic exhibit at least some
irregularity of cardiac rhythm. Arrhythmias are the most important cause of
sudden cardiac death. However, only few arrhythmias need to be treated with
antiarrhythmic drugs.
Abnormal automaticity
or impaired conduction or both underlie cardiac arrhythmias. The generation and
propagation of cardiac impulse and properties of excitability and
refractoriness are described on p. 476 to 478. Ischaemia, electrolyte and pH
imbalance, mechanical injury, stretching, neurogenic and drug influences,
including antiarrhythmics themselves, can cause arrhythmias by altering
electrophysiological properties of cardiac fibres.
Important mechanisms
of cardiac arrhythmias are:
1. Enhanced/Ectopic Pacemaker Activity
The slope of phase4 depolarization may be increased
pathologically in the automatic fibres or such activity may appear in ordinary
fibres. Ectopic impulse may result from current of injury. Myocardial cells
damaged by ischaemia become partially depolarized: a current may flow between
these and normally polarized fibres (injury current) and initiate an impulse.
2. After-Depolarizations
These are secondary
depolarizations accompanying a normal or premature action potential (AP), Fig.
38.1.
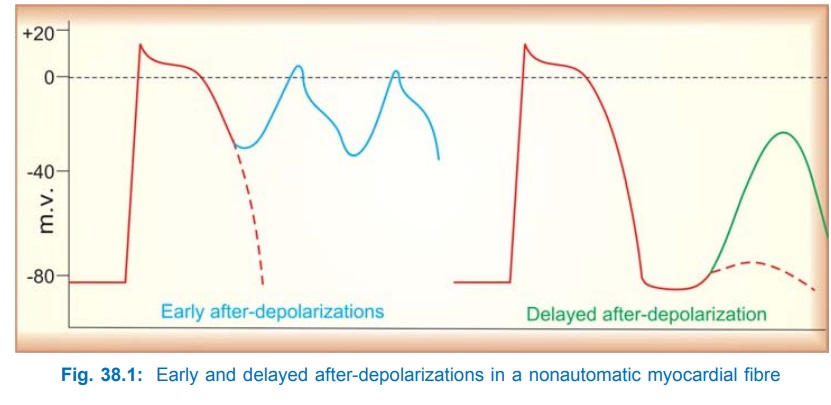
Early After-Depolarization (EAD) Repolarization during phase3 is interrupted and membrane potential
oscillates. If the amplitude of oscillations is sufficiently large,
neighbouring tissue is activated and a series of impulses are propagated. EADs
are frequently associated with long QT interval due to slow repolarization and
prolonged APs. They result from depression of delayed rectifier K+ current.
Delayed After-Depolarization (DAD)
After attaining resting membrane potential
(RMP) a secondary deflection occurs which may reach threshold potential and
initiate a single premature AP. Generally result from Ca2+ overload (digitalis
toxicity, ischaemia-reperfusion).
Because an AP is needed to trigger afterdepolarizations,
arrhythmias based on these have been called triggered
arrhythmias.
3. Reentry
Due primarily to abnormality of conduction, an impulse may recirculate in the heart and
cause repetitive activation without the need for any new impulse to be
generated. These are called reentrant
arrhythmias.
Circus Movement Type: It occurs in an
anatomically defined circuit. A
premature impulse, temporarily blocked in one direction by refractory tissue,
makes a one-way transit around an obstacle (natural orifices in heart,
infarcted or refractory myocardium), finds the original spot in an advanced
state of recovery and re-excites it, setting up recurrent activation of adjacent
myocardium (Fig. 38.2).
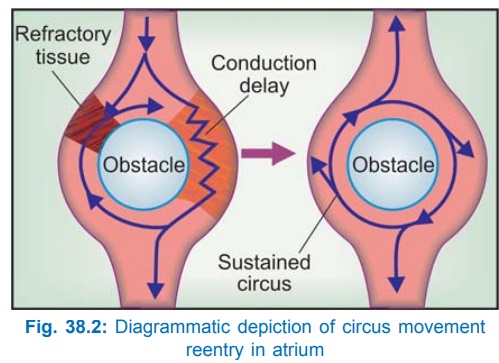
Reentry occurring in an anatomically fixed circuit can be
permanently cured by high radiofrequency catheter ablation of the defined
pathway.
Microreentry
circuit: It may form at the junction
of a Purkinje fibre (PF)
with ordinary ventricular fibre (gate region). One of the branches of the PF
may get sufficiently depolarized to cause unidirectional block (Fig. 38.3).
Extremely slow conduction at this site due to slow channel depolarization and
markedly abbreviated action potential duration (APD) and effective refractory
period (ERP) makes reentry possible in a short loop of tissue.
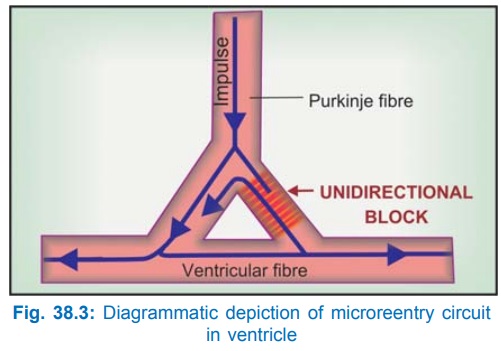
For reentry to occur,
the path length of the circuit should be greater than the wave length (ERP ×
conduction velocity) of the impulse. Slow conduction in the reentrant circuit
may be caused by:
ü Partial depolarization
of the membrane—decreased slope of phase 0 depolarization, i.e. depressed fast
channel response.
ü Cells changing over
from fast channel to slow channel depolarization which conducts extremely
slowly. When a fibre is depolarized to a RMP of about –60 mv, the Na+ (fast)
channels are inactivated, but it can still develop Ca2+ (slow) channel
response.
Reentry can be
abolished both by marked slowing of conduction (converting unidirectional block
to bidirectional block) as well as by acceleration of impulse (retrograde
impulse reaches so early as to meet refractory tissue).
4. Fractionation Of Impulse
When atrial ERP is
brief and inhomogeneous
(under vagal overactivity), an impulse generated early in diastole gets
conducted irregularly over the atrium, i.e. it moves rapidly through fibres
with short ERP (which have completely recovered) slowly through fibres with
longer ERP (partially recovered) and not at all through those still refractory.
Thus, asynchronous activation of atrial fibres occurs → atrial fibrillation
(AF). This arrhythmia must be initiated by a premature depolarization, but is
self sustaining, because passage of an irregular impulse leaves a more
irregular refractory trace and perpetuates the inhomogeneity of ERPs.
The important cardiac
arrhythmias are:
Extrasystoles
(ES) are premature beats due to abnormal
automaticity or afterdepolarization arising from an ectopic focus in the atrium
(AES), AV node (nodal ES) or ventricle (VES). The QRS complex in VES is broader
and abnormal in shape.
Paroxysmal Supraventricular
Tachycardia (PSVT) is sudden onset episodes of atrial tachycardia (rate 150–200/min) with 1:1
atrioventricular conduction: mostly due to circus movement type of reentry
occurring within or around the AV node or using an accessory pathway between
atria and ventricle (Wolff-ParkinsonWhite syndrome).
Atrial Flutter (AFI): Atria beat at a rate
of 200– 350/min and there is a
physiological 2:1 to 4:1 or higher AV block (because AV node cannot transmit
impulses faster than 200/ min). This is mostly due to a stable reentrant
circuit in the right atrium, but some cases may be due to rapid discharge of an
atrial focus.
Atrial fibrillation
(AF): Atrial fibres are activated asynchronously at a rate of
350–550/min (due to electrophysiological inhomogeneity of atrial fibres),
associated with grossly irregular and often fast (100–160/min) ventricular
response. Atria remain dilated and quiver like a bag of worms.
Ventricular Tachycardia is a run of 4 or more consecutive ventricular extrasystoles. It may
be a sustained or non-sustained arrhythmia, and is due either to discharges
from an ectopic focus, afterdepolarizations or single site (monomorphic) or
multiple site (polymorphic) reentry circuits.
Torsades
de pointes (French: twisting of points) is a life-threatening
form of polymorphic ventricular tachycardia with rapid asynchronous complexes
and an undulating baseline on ECG. It is generally associated with long QT
interval.
Ventricular Fibrillation (VF) is grossly irregular, rapid and fractionated activation of
ventricles resulting in incoordinated contraction of its fibres with loss of
pumping function. It is fatal unless reverted within 2–5 min; is the most
common cause of sudden cardiac death.
Atrio-Ventricular (AV) Block is due to depression of impulse conduction through the AV
node and bundle of His, mostly due to vagal influence or ischaemia.
First degree AV block: Slowed conduction resulting in prolonged PR interval.
Second degree AV block: Some supraventricular complexes are not conducted:
drop beats.
Third degree AV block: No supraventricular complexes are conducted; ventricle generates its own impulse;
complete heart block.
Arrhythmogenic Potential Of Antiarrhythmics
Most antiarrhythmics can themselves precipitate
serious arrhythmias, especially during long-term prophylactic use. Two
multicentric trials ‘Cardiac Arrhythmia Suppression Trial I and II’ (CAST I,
II, 1991, 1992) have shown that postMI patients randomized to receive on a long-term
basis encainide, flecainide, moricizine had higher incidence of sudden death,
though initially the same drugs had suppressed VES in these patients. It is
possible that during transient episodes of ischaemia, the intraventricular
conduction slowing action of these drugs gets markedly accentuated resulting in
VT and VF. It is therefore not prudent to try and suppress all
extrasystoles/arrhythmias with drugs.

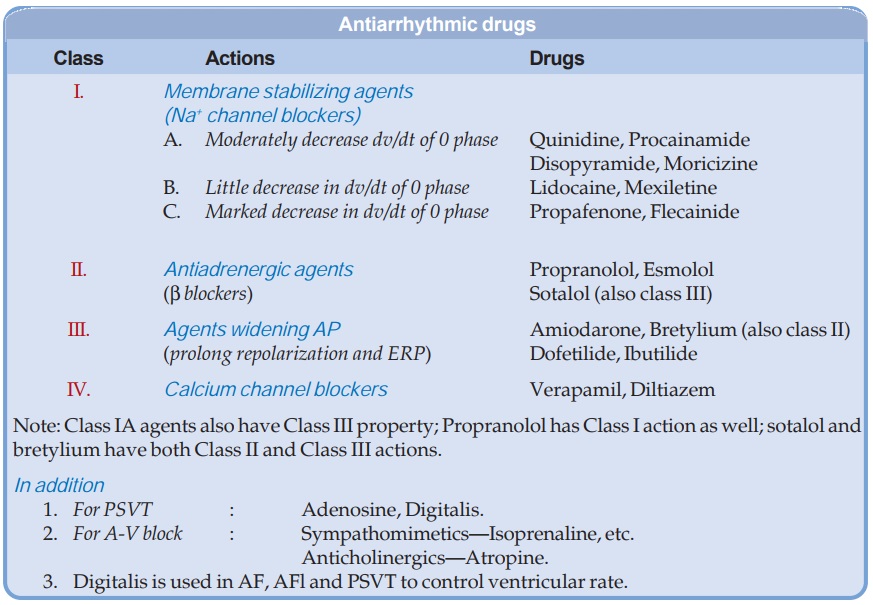
Classification
Antiarrhythmic drugs
act by blocking myocardial Na+, K+ or Ca2+ channels. Some have additional or
even primary autonomic effects. Classification of antiarrhythmic drugs has been
unsatisfactory, because many drugs have more than one action. Vaughan Williams
and Singh (1969) proposed a 4 class system which takes into account the most important
property of a drug which is apparently responsible for its antiarrhythmic
action in the clinical setting. This system, though arbitrary, is widely
accepted.
CLASS I
The primary action of
drugs in this class is to limit the conductance of Na+ (and K+) across cell
membrane—a local anaesthetic action. They also reduce rate of phase-4
depolarization in automatic cells.
SUBCLASS IA
The subclass IA
containing the oldest antiarrhythmic drugs quinidine
and procainamide are open state Na+
channel blockers which also moderately delay channel recovery (1–10s), suppress
AV conduction and prolong refractoriness. The Na+ channel blockade is greater
at higher frequency (premature depolarization is affected more). These actions
serve to extinguish ectopic pacemakers that are often responsible for triggered
arrhythmias and abolish reentry by converting unidirectional block into
bidirectional block.
Quinidine
It is the dextro
isomer of the antimalarial alkaloid quinine found in cinchona bark. In addition
to Na+ channel blockade, quinidine has cardiac antivagal action which augments
prolongation of atrial ERP and minimizes RP disparity of atrial fibres. AV node
ERP is increased by direct action of quinidine, but decreased by its antivagal
action; overall effect is inconsistent. Quinidine depresses myocardial
contractility; failure may be precipitated in damaged hearts.
ECG: It increases PR and QT intervals and tends to broaden QRS complex. Changes in the shape of T
wave may be seen reflecting effect on repolarization.
Mechanism Of Action:
Quinidine blocks
myocardial Na+ channels in the open
state—reduces automaticity and maximal rate of 0 phase depolarization in a
frequency dependent manner. Prolongation of APD is due to K+ channel block,
while lengthening of ERP is caused by its moderate effect on recovery of Na+
and K+ channels. At high concentrations it also inhibits L type Ca2+ channels.
Quinidine decreases the availability of Na+ channels as well as delays their
reactivation.
The other actions of quinidine are fall in BP (weak α adrenergic blockade
and cardiac depression), decreased skeletal muscle contractility, uterine
contractions, vomiting, diarrhoea and neurological effects like ringing in
ears, vertigo, deafness, visual disturbances and mental changes (Cinchonism).
Like its levo isomer, it has antimalarial action, and has been used as a
parenteral alternative to quinine for falciparum malaria. The important drug
interactions of quinidine are:
·
Rise in blood levels and toxicity of digoxin
due to displacement from tissue binding and inhibition of Pglycoprotein
mediated renal and biliary clearance of digoxin.
·
Marked fall in BP in patients receiving
vasodilators.
·
Risk of torsades
de pointes is increased by hypokalaemia caused by diuretics.
·
Synergistic cardiac depression with βblockers, verapamil,
K+ salts.
· Quinidine inhibits CYP2D6: prolongs t½ of
propafenone and inhibits conversion of codeine to morphine.
Use:
Though quinidine is
effective in many atrial and ventricular
arrhythmias, it is not used to terminate them because of risk of adverse
effects, including that of torsades de pointes, sudden cardiac arrest or VF;
idiosyncratic angioedema, vascular
collapse, thrombocytopenia. It is occasionally used in a dose of 100–200 mg TDS
to maintain sinus rhythm after termination of AF or AFI, and rarely in
ventricular arrhythmias.
QUINIDINE SULPHATE 200
mg tab; QUININGA 300 mg tab, 600 mg/2 ml inj; NATCARDINE 100 mg tab.
Procainamide
It is the orally
active amide derivative of the local anaesthetic procaine, with cardiac
electrophysiological actions almost identical to those of quinidine, viz. slowing of 0 phase and impulse
conduction, prolongation of APD, ERP, QRS complex and QT interval. Significant
differences between the two are:
·
It is less effective in suppressing ectopic
automaticity.
·
It causes somewhat less marked depression of
contractility and AV conduction.
·
Antivagal action is minimal.
· It is not an α blocker: causes less
fall in BP; at high doses, fall in BP is due to ganglionic blockade.
Pharmacokinetics
Oral bioavailability
of procainamide is about 75%, peak plasma concentration occurs in 1 hour. It is
metabolized in liver, primarily by acetylation to N-acetyl-procainamide (NAPA)
which has no Na+ channel blocking property but blocks K+ channels and prolongs
repolarization: APD is lengthened. There are fast and slow acetylators of
procainamide (as there are for isoniazid). More than half of procainamide is
excreted unchanged in urine; plasma t½ is relatively short (3–4 hours). Thus,
more frequent dosing than quinidine is required.
Dose: For abolition of arrhythmia—0.5–1 g oral or
i.m. followed by 0.25–0.5 g
every 2 hours; or 500 mg i.v. loading dose (25 mg/min injection) followed by 2
mg/kg/hour. Maintenance dose—0.5 g every 4–6 hours.
PRONESTYL 250 mg tab.,
1 g/10 ml inj.
Adverse Effects
Gastrointestinal
tolerance of procainamide is better
than quinidine, but nausea and vomiting do occur.
CNS: weakness, mental
confusion and hallucinations are noted at higher doses.
Flushing and
hypotension are seen on rapid i.v. injection. Cardiac toxicity, ability to
cause torsades de pointes are similar to quinidine.
Hypersensitivity
reactions are rashes, fever, angioedema. Agranulocytosis and aplastic anaemia
is rare.
More than half of
patients given chronic high dose procainamide therapy develop antinuclear
antibodies and about 1/5 develop systemic lupus erythematosus (SLE). It is more
common in slow acetylators.
Use
Procainamide (i.v.)
can terminate monomorphic VT in upto 80–90% patients, but is less effective in
preventing recurrences. Many WPW reciprocal VTs respond and it has been used to
prevent recurrences of VF. However, procainamide is not suitable for prolonged
oral therapy because of inconveniently frequent dosing and high risk of lupus.
Disopyramide
It is a quinidine like Class IA drug that has prominent cardiac
depressant and anticholinergic actions, but no α adrenergic blocking
property. Disopyramide usually has no effect on sinus rate because of opposing
direct depressant and antivagal actions. Prolongation of PR interval and QRS
broadening are less marked.
Pharmacokinetics
Bioavailability of oral disopyramide is about 80%. It is partly metabolized
in liver by dealkylation, nearly half is excreted unchanged in urine; plasma t½
is 6–8 hrs. The t½ is increased in patients of MI and in renal insufficiency.
Dose: 100–150 mg 6–8 hourly oral.
NORPACE 100, 150 mg
cap, REGUBEAT 100 mg tab.
Adverse Effects
Disopyramide is better
tolerated than quinidine, less g.i. effects. Anticholinergic side effects are
the most prominent: dry mouth, constipation, urinary retention (especially in
elderly males) and blurred vision. It can cause greater depression of cardiac
contractility. Cardiac decompensation and hypotension may occur in patients
with damaged hearts because it also increases peripheral resistance, so that
cardiac output may be markedly decreased.
Contraindications are—sick sinus, cardiac failure and prostate
hypertrophy.
Use
The primary indication
of disopyramide is as a second line drug
for prevention of recurrences of ventricular arrhythmia. It may also be used
for maintenance therapy after cardioversion of AF or AFl.
Moricizine
This Class IA drug delays Na+ channel recovery to a greater extent (also classified
as Class IC), but cardio-depressant and CNS effects are less marked. It has
been used to suppress VES and WPW arrhythmias, but the CAST II study has found
it to increase mortality in postMI patients.
SUBCLASS IB
These drugs block Na+
channels more in the inactivated than in the open state, but do not delay
channel recovery (channel recovery time < 1S). They do not depress AV
conduction or prolong (even shorten) APD, ERP and QT.
Lidocaine (Lignocaine)
It is the most
commonly used local anaesthetic. In addition, it is a popular antiarrhythmic in
intensive care units.
The most prominent
cardiac action of lidocaine is suppression of automaticity in ectopic foci.
Enhanced phase4 depolarization in partially depolarized or stretched PFs, and
afterdepolarizations are antagonized, but SA node automaticity is not
depressed.
The rate of 0 phase
depolarization and conduction velocity in AV bundle or ventricles is not
decreased. Lidocaine decreases APD in PF and ventricular muscle, but has
practically no effect on APD and ERP of atrial fibres. Atrial reentry is not
affected. However, it can suppress reentrant ventricular arrhythmias either by
abolishing one-way block or by producing two way block.
Lidocaine is a blocker
of inactivated Na+ channels more than that of open state. As such, it is
relatively selective for partially depolarized cells and those with longer APD
(whose Na+ channels remain inactivated for longer period). While normal ventricular
and conducting fibres are minimally affected, depolarized/damaged fibres are
significantly depressed. Brevity of atrial AP and lack of lidocaine effect on
channel recovery might explain its inefficacy in atrial arrhythmias.
Lidocaine has minimal
effect on normal ECG; QT interval may decrease. It causes little depression of
cardiac contractility or arterial BP. There are no significant autonomic
actions: all cardiac effects are direct actions.
Pharmacokinetics
Lidocaine is inactive
orally due to high first pass
metabolism in liver. Action of an i.v. bolus lasts only 10–20 min because of
rapid redistribution. It is hydrolysed, deethylated and conjugated; metabolites
are excreted in urine. Metabolism of lidocaine is hepatic blood flow dependent.
The t½ of early
distribution phase is 8 min while that of later elimination phase is nearly 2
hours. Its t½ is prolonged in CHF, because of decrease in volume of
distribution and hepatic blood flow.
Dose and preparations Lidocaine is given
only by i.v. route: 50–100 mg bolus
followed by 20–40 mg every 10– 20 min or 1–3 mg/min infusion.
XYLOCARD, GESICARD 20 mg/ml inj. (5, 50 ml vials). These preparations for
cardiac use contain no preservative. The local anaesthetic preparations should
not be used for this purpose.
Ventricular ectopic activity can be titrated with the rate of
administration. Propranolol prolongs t½ of lidocaine by reducing hepatic blood
flow. Cimetidine also increases plasma levels of lidocaine.
Adverse Effects
The main toxicity is dose
related neurological effects:
Drowsiness, nausea, paresthesias, blurred vision, disorientation,
nystagmus, twitchings and fits. Lidocaine has practically no proarrhythmic
potential and is the least cardiotoxic antiarrhythmic. Only excessive doses cause
cardiac depression and hypotension.
Use
Lidocaine is used only
in ventricular tachyarrhythmias. It is ineffective in atrial arrhythmias.
Because of rapidly developing and titratable action it is a good drug in the
emergency setting, e.g. arrhythmias following acute MI, during cardiac surgery,
etc. Given prophylactically by infusion in acute MI, it reduces occurrence of
VF. However, metaanalysis has shown that lidocaine fails to improve survival;
may even increase short term mortality. Therefore, it is no longer administered
routinely to all MI patients.
Efficacy of lidocaine in chronic ventricular arrhythmia is low,
but it is useful in digitalis toxicity because it does not worsen AV block.
Mexiletine
It is a local anaesthetic and an active antiarrhythmic by the
oral route; chemically and pharmacologically similar to lidocaine. It reduces
automaticity in PF, both by decreasing phase4 slope and by increasing threshold
voltage. By reducing the rate of 0 phase depolarization in ischaemic PF it may convert
oneway block to twoway block.
Mexiletine is almost completely absorbed orally, 90% metabolized
in liver and excreted in urine; plasma t½ 9–12 hours.
Bradycardia,
hypotension and accentuation of AV block may attend i.v. injection of
mexiletine. Neurological—tremor, nausea and vomiting are common; dizziness,
confusion, blurred vision, ataxia can occur.
Dose: 100–200 mg i.v. over
10 min., 1 mg/min infusion.
Oral: 150–200 mg TDS
with meals.
MEXITIL 50, 150 mg
caps, 250 mg/10 ml. inj.
Use
Parenteral mexiletine
is effective in postinfarction ventricular arrhythmias as alternative to
lidocaine in resistant cases. Orally it is used to keep VES and VT suppressed
over long-term.
SUBCLASS IC
These are the most
potent Na+ channel blockers with more prominent action on open state and the
longest recovery times (> 10S). They markedly delay conduction, prolong PR,
broaden QRS complex, but have variable effect on APD. Drugs of this subclass
have high proarrhythmic potential—sudden deaths have occurred.
They have profound
effect on His-Purkinje as well as accessory pathway conduction; markedly retard
anterograde as well as retrograde conduction in the bypass tract of WPW
syndrome.
Propafenone
By blocking Na+ channels propafenone markedly
depresses conduction and has β adrenergic blocking property—can precipitate
CHF and bronchospasm. Sinoatrial block has occurred occasionally. Propafenone
is absorbed orally and undergoes variable first pass metabolism; there being extensive or poor metabolizers. Bioavailability and t½ differs considerably among
individuals. Some metabolites are active. Side effects are nausea, vomiting,
bitter taste, constipation and blurred vision.
Propafenone is a
reserve drug for ventricular arrhythmias, reentrant tachycardias involving AV
node/accessory pathway and to maintain sinus rhythm in AF.
Dose: 150 mg BD–300 mg TDS; RHYTHMONORM 150 mg
tab.
Flecainide
It suppresses VES, VT,
WPW tachycardia and prevents
recurrences of AF and PSVT. But in the CAST study it was found to increase
mortality in patients recovering from MI; can itself provoke arrhythmias during
chronic therapy. It is reserved for resistant cases of recurrent AF, and WPW
rhythms in patients not having associated CHF.
CLASS II
The primary action of
drugs in this class is to suppress adrenergically mediated ectopic activity.
Propranolol
Some β blockers, e.g. propranolol have quinidine like direct
membrane stabilizing action at high doses, but in the clinically used dose
range—antiarrhythmic action is exerted primarily because of cardiac adrenergic
blockade. Propranolol decreases the slope of phase4 depolarization and
automaticity in SA node, PF and other ectopic foci when this has been increased
under adrenergic influence; little action otherwise. The other most important
action is to prolong the ERP of AV node (an antiadrenergic action). This impedes
AV conduction (no paradoxical tachycardia can occur when atrial rate in AF or
AFl is reduced).
Slow channel responses
and afterdepolarizations that have been induced by catecholamines (CAs) are suppressed.
Reentrant arrhythmias that involve AV node (many PSVTs) or that are dependent
on slow channel/depressed fast channel response may be abolished by its marked
depressant action on these modalities.
The most prominent ECG
change is prolongation of PR interval. Depression of cardiac contractility and
BP are less marked than with quinidine.
Administration
For rapid action,
propranolol may be injected i.v. 1 mg/min
(max. 5 mg) under close monitoring. The usual oral antiarrhythmic dose is 40–80
mg 2–4 times a day.
Use
Propranolol is very
useful in treating inappropriate sinus
tachycardia, atrial and nodal ESs provoked by emotion or exercise. It is less
effective than adenosine and verapamil for PSVT—conversion rate is about 60%.
Propranolol rarely abolishes
AF or AFl, but can be used to control ventricular rate. It is highly effective
in sympathetically mediated arrhythmias seen in pheochromocytoma and during
anaesthesia with halothane. Digitalis induced tachyarrhythmias may be
suppressed.
Efficacy in chronic
ventricular arrhythmias is low, but its anti-ischaemic action may be
protective. Prophylactic treatment with β blockers reduces
mortality in postMI patients. Propranolol has also been used for WPW, but in
some cases severe bradycardia may be precipitated.
Sotalol
It is a nonselective β blocker having prominent Class III action of
prolonging repolarization by blocking cardiac K+ channels. It is not a Na+
channel blocker—does not depress conduction in fast response tissue, but delays
AV conduction and prolongs its ERP. Sotalol is effective in polymorphic VT and
for maintaining sinus rhythm in AF/AFl. Due to prolongation of APD and QT, risk
of dose-dependent torsades de pointes is the major limitation. It is
contraindicated in patients with long
QT interval.
Esmolol
This quick and short
acting β1 blocker administered
i.v. is very useful for emergency control of ventricular rate in
AF/AFl. It can terminate supraventricular tachycardia, and is mainly used for
arrhythmias associated with anaesthesia.
MINIBLOCK 100 mg/10
ml, 250 mg/10 ml inj.; 0.5 mg/ kg in 1 min followed by 0.05–0.2 mg/kg/min i.v.
infusion.
CLASS III
The characteristic
action of this class is prolongation of repolarization; AP is widened and ERP
is increased. The tissue remains refractory even after full repolarization:
reentrant arrhythmias are terminated.
Amiodarone
This unusual iodine
containing highly lipophilic long-acting antiarrhythmic exerts multiple
actions:
Prolongs APD and QT interval attributable to block of myocardial
delayed rectifier K+ channels. This also appears to reduce nonuniformity of
refractoriness among different fibres.
Preferentially blocks
inactivated Na+ channels (like lidocaine) with relatively rapid rate of channel
recovery: more effective in depressing conduction in cells that are partially
depolarized or have longer APD.
Inhibits myocardial
Ca2+ channels and has noncompetitive β adrenergic blocking property.
Conduction is slowed and ectopic automaticity is markedly
depressed, but that of SA node is affected only slightly. Effect of oral doses
on cardiac contractility and BP are minimal, but i.v. injection frequently
causes myocardial depression and hypotension.
Despite prolongation
of APD, the arrhythmia (torsades de pointes) provoking potential of
amiodarone is low, probably because
it does not exhibit ‘reverse use-dependence’ of APD prolongation or because of
its multiple antiarrhythmic mechanisms. The prolongation of APD by most class
III drugs is more marked at slower rates of activation (encouraging EAD) than
at higher rates (reverse use-dependence), while with amiodarone it is
independent of rate of activation.
Pharmacokinetics
Amiodarone is incompletely and slowly absorbed
from the g.i.t. On daily oral ingestion the action develops over several days, even
weeks. However, on i.v. injection, action develops rapidly. It accumulates in
muscle and fat from which it is slowly released and then metabolized in liver
mainly by CYP3A4. One metabolite is active. The duration of action is
exceptionally long; t½ 3–8 weeks.
Dose: Amiodarone is mainly used orally 400–600 mg/ day for few weeks, followed by 100–200 mg OD
for maintenance therapy. 100–300 mg (5 mg/kg) slow i.v. injection over 30–60
min.
CORDARONE, ALDARONE, EURYTHMIC 100, 200 mg tabs, 150 mg/3 ml
inj.
Use
Amiodarone has been
found effective in a wide range of
ventricular and supraventricular arrhythmias. Resistant VT and recurrent VF are
the most important indications. It is also used to maintain sinus rhythm in AF
when other drugs have failed. Rapid termination of ventricular and
supraventricular arrhythmias can be obtained by i.v. injection. WPW
tachyarrhythmia is terminated by suppression of both normal and aberrant
pathways.
Its long duration of
action makes it suitable for long-term prophylactic therapy; has been found to
reduce sudden cardiac death. Because of high and broad spectrum efficacy and
relatively low proarrhythmic potential, amiodarone is a commonly used
antiarrhythmic, despite its organ toxicity in the long-term.
Adverse Effects
These are dose-related
and increase with duration
of therapy. Fall in BP, bradycardia and myocardial depression occurs on i.v. injection
and on drug cumulation.
Nausea,
gastrointestinal upset may attend oral medication, especially during the
loading phase. Photosensitization and skin pigmentation occurs in about 10%
patients. Corneal microdeposits are common with long-term use, but are reversible
on discontinuation.
Pulmonary alveolitis
and fibrosis is the most serious toxicity of prolonged use, but is rare if daily
dose is kept below 200 mg.
Peripheral neuropathy
generally manifests as weakness of shoulder and pelvic muscles. Liver damage is
rare. Amiodarone interferes with thyroid function in many ways including
inhibition of peripheral conversion of T4 to T3: goiter,
hypothyroidism and rarely hyperthyroidism may develop on chronic use.
Interactions
Amiodarone can
increase digoxin and warfarin levels by
reducing their renal clearance. Additive AV block can occur in patients
receiving β blockers or calcium
channel blockers. Inducers and inhibitors of CYP3A4 respectively decrease and
increase amiodarone levels.
Bretylium
It is an adrenergic
neurone blocking drug (see Ch. No. 10) introduced in 1960 as
antihypertensive, but was soon withdrawn. It was reintroduced for parenteral
use to facilitate reversal of VF, but is not available in India or the USA, and
is rarely used elsewhere.
Bretylium has complex
electrophysiological effects which are partly a result of initial NA release
from adrenergic terminals in heart, and later blockade of NA release, but major
direct action is prolongation of APD and ERP, due to K+ channel blockade.
Dofetilide
This newer antiarrhythmic prolongs APD and ERP by selectively blocking rapid component of
delayed rectifier K+ current without affecting other channels or receptors; has
no autonomic or peripheral actions. It is therefore labelled as pure class III antiarrhythmic.
Oral dofetilide can convert AF or AFl to sinus rhythm in ~30%
cases, but is more effective in maintaining sinus rhythm in converted
patients—its primary indication. Significantly, chronic therapy with dofetilide
in patients with high risk of sudden cardiac death/post MI cases has not
increased mortality, despite provoking torsades
de pointes in some recipients. It
is mainly excreted unchanged in urine
and produces few side effects.
Ibutilide is another new class
III antiarrhythmic used i.v. for pharmacological
conversion of AFl and AF to sinus rhythm.
CLASS IV
The primary action of this class of drugs is to inhibit Ca2+
mediated slow channel inward current.
Verapamil
Of the many Ca2+ channel blockers, verapamil has the most
prominent cardiac electrophysiological action (Table 38.1). It blocks L type
Ca2+ channels and delays their recovery. Its antiarrhythmic aspects are
described here, while other aspects are covered in Ch. No. 39 and 40.
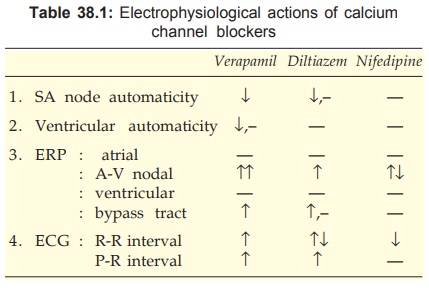
The basic action of
verapamil is to depress Ca2+ mediated depolarization. This suppresses automaticity
or reentry dependent on slow response. Phase4 depolarization in SA node and PFs
is reduced resulting in bradycardia and extinction of latent pacemakers. Reflex
sympathetic stimulation due to vasodilatation partly counteracts the direct bradycardia
producing action. Delayed afterdepolarizations in PFs are dampened.
The most consistent
action of verapamil is prolongation of AV nodal ERP. As a result AV conduction
is markedly slowed and reentry involving AV node is terminated. Intraventricular
conduction, however, is not affected. Verapamil has negative inotropic action
due to interference with Ca2+ mediated excitation-contraction coupling in
myocardium.
Uses and Precautions
ü PSVT—Verapamil can terminate
attacks of PSVT; 5 mg i.v. over 2–3 min is effective in 80% cases, but marked
bradycardia, AV block, cardiac arrest and hypotension can occur. Verapamil
should not be used if PSVT is accompanied with hypotension or CHF. It is also
useful for preventing recurrences: 60 to 120 mg TDS orally.
ü To control ventricular
rate in AF or AFl; it may be used as an alternative to, or in addition to
digitalis: 40–80 mg TDS oral. In few cases the AF or AFl may revert to sinus
rhythm, but this is an unusual happening.
Reentrant
supraventricular and nodal arrhythmias (WPW) are susceptible to verapamil, but
it should not be used because of risk of increased ventricular rate due to
reflex sympathetic stimulation and reduction of ERP of the bypass tract in some
cases.
Verapamil has poor
efficacy in ventricular arrhythmias. In contrast to β blockers, verapamil
prophylaxis does not reduce mortality in post-MI patients. In some patients of
VT, i.v. injection of verapamil has precipitated VF: therefore contraindicated.
It is also not recommended for digitalis toxicity, because additive AV block may
occur. It is contraindicated in partial heart block and sick sinus.
CALAPTIN 40, 80 mg
tab; 120, 240 mg SR tab, 5 mg/2 ml inj.
Diltiazem
The direct cardiac actions of diltiazem are similar to those of verapamil. However, they
are less marked. It is an alternative to verapamil for PSVT.
For rapid control of ventricular rate in AF or AFl, i.v.
diltiazem is preferred over verapamil, because it can be more easily titrated
to the target heart rate, causes less hypotension and myocardial depression—can
be used even in the presence of mild-to-moderate CHF.
DILZEM 30, 60, 90 mg
tabs, 25 mg/5 ml inj.
Related Topics
