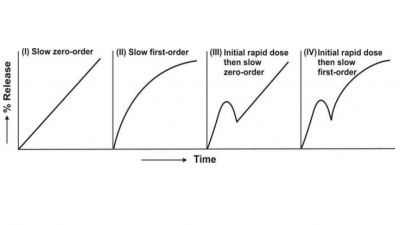
Classification of CRDDS
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Controlled Release Medication
CRDDS can be classified in various ways – 1. On the basis of technical sophistication 2. On the basis of route of administration.
CLASSIFICATION OF CRDDS
CRDDS can be classified in various ways –
1. On the basis of technical
sophistication
2. On the basis of route of
administration.
On the basis of technical sophistication, CRDDS can
be categorised into 4 major classes:
1. Rate-programmed DDS
2. Activation-controlled DDS
3. Feedback-controlled DDS
4. Site-targeted DDS
In the former three cases i.e. except site-targeted
DDS, the formulation comprise of three basic components –
i. The drug
ii. The rate controlling element
iii. Energy source that activates
the DDS.
1. Rate-Programmed DDS
These DDS are those from which the drug release has
been programmed at specific rate profiles. They are further subdivided into
following subclasses:
i.
Dissolution-controlled DDS
ii.
Diffusion-controlled DDS
iii.
Dissolution and
diffusion-controlled DDS.
All the above systems can be designed in one of the
following ways –
i.
Reservoir systems
(membrane-controlled DDS)
ii.
Matrix systems
(soluble/erodible/swellable/degradable)
iii.
Hybrid systems (i.e. membrane cum
matrix systems)
1. Dissolution-Controlled DDS
These systems are those where the rate-limiting
phenomenon responsible for imparting the controlled-release characteristics to
the DDS is either of the two -
(a) Slow dissolution rate of the
drug - the drug present in such a system may be one of the
following two types:
i. Drug with inherently slow
dissolution rate e.g. griseofulvin, digoxin and nifedipine.
Such drugs act as natural prolonged-release products, or
ii. Drug that transforms into
slow dissolving forms on contact with GI fluids e.g. ferrous
sulphate.
(b) Slow dissolution rate of the
reservoir membrane or matrix - the drug
present in such a system may be the one having high aqueous solubility and
dissolution rate e.g. pentoxifylline and metformin. The challenge in designing
such systems lies in controlling the drug dissolution rate by employing either or
combination of following techniques –
i. Embedment in slowly
dissolving, degrading or erodible matrix. The matrix in addition may have low
porosity or poor wettability.
ii. Encapsulation or coating with
slow-dissolving, degrading or erodible substances. In this approach, the rate
of dissolution fluid penetration and/or wettability of the reservoir system are
controlled.
Slowly soluble and erodible materials commonly
employed to achieve these objectives include hydrophobic substances such as
ethyl cellulose (containing an added water-soluble release modifying agent such
as PVP), polymethacrylates with pH independent solubility (e.g. Eudragit RS and
RL 100) and waxes such as glyceryl monostearate, and hydrophilic materials like
sodium CMC.
2. Diffusion-Controlled DDS
These systems are those where the rate-controlling
step is not the dissolution rate of drug or release controlling element, but
the diffusion of dissolved drug molecule through the rate-controlling element.
The rate-controlling element in such a system is thus neither soluble, erodible
nor degradable but is water-swellable or water-insoluble. Water-swellable
materials include hydrophilic polymers and gums such as xanthan gum, guar gum,
high viscosity grades of HPMC and HPC, alginates, etc. Water-insoluble polymers
most commonly used in such systems are ethyl cellulose and polymethacrylates.
3. Dissolution and Diffusion-Controlled DDS
These systems are those where the rate of drug
release is controlled by drug or polymer dissolution as well as drug diffusion
i.e. the system is a combination of the two systems discussed above.
A summary of various approaches that are employed
in the design of rate-programmed DDS is illustrated in figure 14.6.
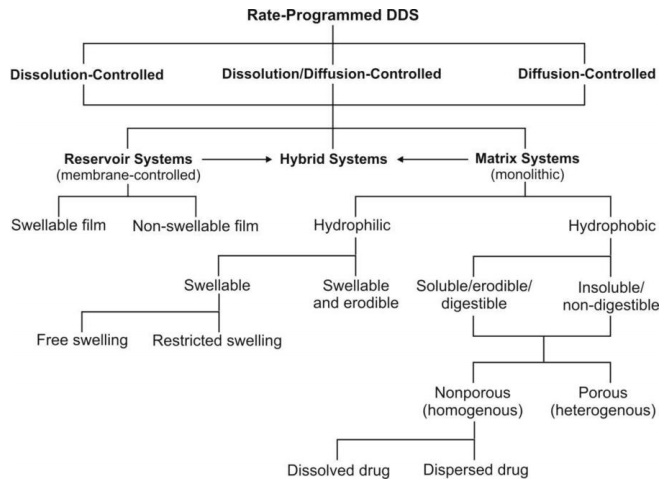
Fig. 14.6 Approaches in the design of rate-programmed DDS
i. Reservoir systems
(membrane-controlled DDS) – (see also Modern Pharmaceutics for
advantages and disadv) These systems are those
where the drug is present as a core in a compartment of specific shape encased or
encapsulated with a rate-controlling wall, film or membrane having a
well-defined thickness. The drug in the core must dissociate themselves from
the crystal lattice and dissolve in the surrounding medium, partition and
diffuse through the membrane.
Depending upon the physical properties of the
membrane, two types of reservoir systems are possible –
(a) Non-swelling reservoir systems – are those where the polymer membrane do
not swell or hydrate in aqueous
medium. Ethyl cellulose and polymethacrylates are commonly used polymers in
such systems. Such materials control drug release owing to their thickness,
insolubility or slow dissolution or porosity. Reservoir system of this type is
most common and includes coated drug particles, crystals, granules, pellets,
minitablets and tablets.
(b) Swelling-controlled reservoir systems – are those where the polymer
membrane swell or hydrate on contact
with aqueous medium. In such systems drug release is delayed for the time
period required for hydration of barrier and after attainment of barrier
hydration, drug release proceeds at a constant rate. HPMC polymers are commonly
employed in such systems.
ii. Matrix systems (monolithic
DDS) – (see
also Modern Pharmaceutics for advantages and
disadv) These systems are those where the drug
is uniformly dissolved or dispersed in release-retarding material. Such devices can
be formulated as conventional matrix, or bi-or tri-layered matrix systems.
Depending upon the physical properties of the
membrane, two types of matrix devices are possible –
(a) Hydrophilic matrix – is the one where the release retarding
material is a water swellable or
swellable cum erodible hydrocolloid such as high molecular weight HPMCs, HPC,
HEC, xanthan gum, sodium alginate, guar gum, locust bean gum, PEO (polyethylene
oxide) and cross linked polymers of acrylic acid. Hydrophilic matrices are
porous systems.
Depending upon the swelling behaviour of
hydrophilic polymer, two types of matrices are possible –
·
Free-swelling matrix – is the one in which swelling is
unhindered.
·
Restricted-swelling matrix – is the
one in which the surface of the device is
partially coated with an impermeable polymer film that restricts the
hydration of swellable matrix material.
(b) Hydrophobic matrix – is the one where the release retarding
material is either –
·
Slowly soluble, erodible or digestible, for
e.g. waxes such as glyceryl monostearate,
cetyl alcohol, hydrogenated vegetable oils, beeswax, carnauba wax, etc.
·
Insoluble or non-digestible, for e.g.
ethyl cellulose, polymethacrylates, etc.
Depending upon the manner of incorporation of drug
in the matrix, hydrophobic matrices can be further classified as –
a. Porous (heterogeneous) matrix – is the one where the drug and
release-retarding matrix microparticles are simply mixed with each other and compressed
into a tablet or the drug is dispersed in the polymer solution followed by
evaporation of the solvent.
b. Nonporous (homogeneous) matrix – is the
one in which the release-retarding matrix
material is first melted and the drug is then incorporated in it by thorough
mixing followed by congealing the mass while stirring. Two types of nonporous
matrix systems are possible –
·
Dissolved drug nonporous system – is the one where the drug is dissolved in the molten
release-retarding matrix material.
·
Dispersed drug nonporous system – is the one where the quantity of drug is greater than its solubility
in molten matrix polymer.
iii. Hybrid systems (membrane cum
matrix DDS) – (see
also Modern Pharmaceutics for advantages
and disadv) These systems are those where the
drug in matrix of release-retarding material is further coated with a
release-controlling polymer membrane. Such a device thus combines the constant
release kinetics of reservoir system with the mechanical robustness of matrix
system.
2. Activation-Controlled DDS
In this group of CRDDSs, the release of drug
molecules from the delivery systems is activated by some physical, chemical, or
biochemical processes and/or facilitated by an energy supplied externally (Fig. 2 Chien article).
The rate of drug release is then controlled by regulating the process applied
or energy input. Based on the nature of the process applied or the type of
energy used, these activation-controlled DDSs can be classified into following
categories:
A. Activation by Physical
Processes
1. Osmotic pressure-activated DDS
2. Hydrodynamic
pressure-activated DDS
3. Vapour pressure-activated DDS
4. Mechanical force-activated DDS
5. Magnetically-activated DDS
6. Sonophoresis-activated DDS
7. Iontophoresis-activated DDS
B. Activation by Chemical
Processes
1. pH-activated DDS
2. Ion-activated DDS
3. Hydrolysis-activated DDS
C. Activation by Biochemical
Processes
1. Enzyme-activated DDS
A. Physical Process-Activated DDS
1. Osmotic Pressure-Activated DDS
Osmotic systems release drug at a predetermined,
typically zero-order rate, based on the principle of osmosis. Osmosis is
natural movement of a solvent through a semipermeable membrane into a solution
of higher solute concentration, leading to equal concentration of the solute on
either sides of the membrane. Osmotic systems imbibe water from the body
through a semipermeable membrane into an osmotic material which dissolves in it
and increase in volume and generate osmotic pressure that results in slow and
even delivery of drug through an orifice.
A semipermeable membrane (e.g. cellulose acetate)
is the one that is permeable to a solvent (e.g. water) but impermeable to ionic
(e.g. sodium chloride) and high molecular weight compounds.
In comparison to DDS based on diffusion and
erosion, osmotic systems are more complex in design but provide better
zero-order drug delivery.
2. Hydration/Hydrodynamic Pressure-Activated DDS
These systems are identical to osmotic systems that
release drug at a zero-order rate. It however differs from osmotic system in
that hydrodynamic pressure generating agent which is typically a water
swellable hydrocolloid such as HPMC is contained in one compartment and the
drug solution/dispersion in another collapsible reservoir. Both these
compartments are housed in a rigid, shape retaining but water permeable
housing. The hydrocolloid imbibes water and swells to generate hydrodynamic
pressure that pushes the drug reservoir compartment and thus force the drug
through an orifice at a slow and uniform rate.
3. Vapour Pressure-Activated DDS
These systems are identical to hydrodynamic systems
in that the pumping compartment and the drug solution/dispersion compartment
are separated by a freely movable partition and the whole system is enclosed in
a rigid housing. The pumping compartment contains a liquefied compressed gas
that vaporises at body temperature and creates vapour pressure that moves the
partition to force the drug out of the device through a series of flow
regulator and delivery cannula into the blood circulation at a constant rate. A
typical example is the development infusion pump of heparin in anticoagulant
therapy, of insulin in the control of diabetes and of morphine for patients
suffering from the intensive pain of a terminal cancer.
4. Mechanical Force-Activated DDS
In these systems the drug reservoir is a solution
in a container equipped with a mechanically activated pumping system. A metered
dose of drug formulation can be reproducibly delivered into a body cavity, such
as the nose, through the spray head upon manual activation of the drug-delivery
pumping system. The volume of solution delivered is fixed and is independent of
the force and duration of activation. A
typical example of this type of drug-delivery system is the development of a
metered-dose nebuliser for the intranasal administration of a precision dose of
luteinizing hormone-releasing hormone (LHRH) and its synthetic analogues, such
as buserelin.
5. Magnetically-Activated DDS
In these systems a tiny doughnut-shaped magnet is
positioned in the centre of a hemispherical shaped drug-dispersing
biocompatible polymer matrix and then coating the external surface of the
medicated polymer matrix, with the exception of one cavity at the centre of the
flat surface of the hemisphere, with a pure polymer, for instance, ethylene–
vinyl acetate copolymer or silicone elastomers. This uncoated cavity is
designed for allowing a peptide drug to release. When the magnet is activated,
to vibrate by an external electromagnetic field, it releases the drug at a
zero-order rate by diffusion process. (fig in Chien)
6. Sonophoresis-Activated DDS
This type of activation-controlled drug delivery
system utilizes ultrasonic energy to activate or trigger the delivery of drugs
from a polymeric drug delivery device. The system can be fabricated from either
a non-degradable polymer, such as ethylene–vinyl acetate copolymer, or a
bioerodible polymer, such as poly(lactide–glycolide) copolymer.
7. Iontophoresis-Activated DDS
This type of CRDDS uses electrical current to
activate and modulate the diffusion of a charged drug molecule across a
biological membrane, such as the skin, in a manner similar to passive diffusion
under a concentration gradient but at a much faster rate. It is a painless
procedure. Since like charges repel each other, application of a positive
current drives positively charged drug molecules away from the electrode and
into the tissues; and vice versa. (see fig. In book
rev, what is iontophoresis, absorption of drug folder)
A typical example of this type of
activation-controlled system is percutaneous penetration of anti-inflammatory
drugs such as dexamethasone to surface tissues.
B. Chemical Process-Activated DDS
1. pH-Activated DDS
These systems are designed for acid-labile drugs or
drugs irritating to gastric mucosa and target their delivery to the intestinal
tract. It is fabricated by coating a core tablet of such a drug with a
combination of intestinal fluid-insoluble polymer, like ethyl cellulose, and
intestinal fluid-soluble polymer, like HPMCP. In the stomach, the coating
membrane resists dissolution in pH 1-3. After gastric emptying, the system
travels to the small intestine, and the intestinal fluid-soluble component in
the coating membrane is dissolved in at pH above 5 thereby producing a
microporous membrane that controls the release of drug from the core tablet. An
example of such a system is oral controlled delivery of potassium chloride,
which is highly irritating to gastric epithelium. (fig.
Chien)
2. Ion-Activated DDS
Based on the principle that the GIT has a
relatively constant level of ions, this type of system has been developed for
controlling the delivery of an ionic or an ionisable drug at a constant rate.
Such a CRDDS is prepared by first complexing an ionisable drug with an
ion-exchange resin. A cationic drug is complexed with a resin containing SO3−
group or an anionic drug with a resin containing N(CH3)3+
group. The granules of the drug–resin complex are further treated with an
impregnating agent, like polyethylene glycol 4000, for reducing the rate of
swelling upon contact with an aqueous medium. They are then coated by an
air-suspension coating technique with a water-insoluble but water-permeable
polymeric membrane, such as ethyl cellulose. This membrane serves as a
rate-controlling barrier to modulate the release of drug from the CRDDS. In the
GI tract, hydronium and chloride ions diffuse into the CRDDS and interact with
the drug–resin complex to trigger the dissociation and release of ionic drug
Acidic drug release
Basic drug release
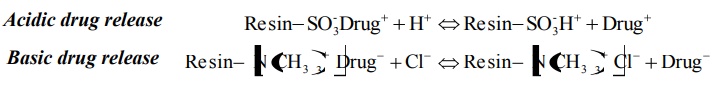
An example of such a formulation is liquid-oral
with sustained release of a combination of hydrocodone and chlorpheniramine. (Fig. Chien).
3. Hydrolysis-Activated DDS
This type of CRDDS depends on the hydrolysis
process to activate the release of drug molecules. In this system, the drug
reservoir is either encapsulated in microcapsules or homogeneously dispersed in
microspheres or nanoparticles prepared from bioerodible or biodegradable
polymers such as polylactide, poly(lactide–glycolide) copolymer,
poly(orthoester) or poly(anhydride). The release of a drug from the polymer
matrix is activated by the hydrolysis-induced degradation of polymer chains,
and the rate of drug delivery is controlled by polymer degradation rate. A
typical example is injectable microspheres for the subcutaneous controlled delivery
of luprolide, a potent biosynthetic analogue of gonadotropin-releasing hormone
(GnRH) for the treatment of gonadotropin-dependent cancers, such as prostate
carcinoma in men and endometriosis in the females, for up to 4 months. (Fig. Chien).
C. Biochemical Process-Activated DDS
1. Enzyme-Activated DDS
In this type of CRDDS, the drug reservoir is either
physically entrapped in microspheres or chemically bound to polymer chains
fabricated from biopolymers, such as albumins or polypeptides. The release of
drugs is made possible by the enzymatic hydrolysis of biopolymers by a specific
enzyme in the target tissue. A typical example is the development of albumin
microspheres, which release 5-fluorouracil, in a controlled manner, by
protease-activated biodegradation.
3. Feedback-Controlled DDS
In this group of CRDDSs, the release of drug
molecules is activated by a triggering agent, such as a biochemical substance,
in the body via some feedback mechanisms. The rate of drug release is regulated
by the concentration of a triggering agent detected by a sensor built into the
CRDDS. (fig chien)
1. Bioerosion-Activated DDS
This CRDDS consists of drug dispersed in a
bioerodible matrix made of poly(vinyl methyl ether) half-ester, which is coated
with a layer of immobilized urease. In a solution at neutral pH, the polymer
erodes slowly. In the presence of urea, urease at the surface of the drug delivery
system metabolizes urea to form ammonia. This causes the pH to increase which
activates a rapid degradation of polymer matrix and subsequently release of
drug molecules.
2. Bioresponsive DDS
In this CRDDS, the drug reservoir is contained in a
device enclosed by a bioresponsive polymeric membrane whose permeability to
drug molecules is controlled by the concentration of a biochemical agent in the
tissue where the CRDDS is located. A typical example of this is the development
of a glucose-triggered insulin delivery system, in which the insulin reservoir
is encapsulated within a hydrogel membrane containing pendant NR2
groups. In an alkaline solution, the NR2 groups exist at neutral
state and the membrane is not swollen and thus impermeable to insulin. As
glucose penetrates into the membrane, it is oxidized enzymatically by the
glucose oxidase entrapped in the membrane to form gluconic acid. This process
triggers the protonation of NR2 groups to form NR2H+,
and the hydrogel membrane becomes swollen and permeable to insulin molecules.
The amount of insulin delivered is bioresponsive to the concentration of
glucose penetrating into the CRDDS. (fig chien)
3. Self-Regulating DDS
This type of feedback-controlled DDS depends on a
reversible and competitive binding mechanism to activate and to regulate the
release of drug. The drug reservoir is a drug complex encapsulated within a
semipermeable polymeric membrane. The release of drug from the CRDDS is
activated by the membrane permeation of a biochemical agent from the tissue
where the CRDDS is located. An example of this is development of
self-regulating insulin delivery system that utilizes complex of glycosylated
insulin– concanavalin A, which is encapsulated inside a polymer membrane. As
glucose penetrates into the system, it activates the release of glycosylated
insulin from the complex for a controlled release from the system. The amount
of insulin released is thus self-regulated by the concentration of glucose that
has penetrated into the insulin delivery system.
4. Site-Targeted DDS
Most conventional dosage forms deliver drug into
the body that eventually reaches the site of action by multiple steps of
diffusion and partitioning. In addition to the target site, the drug also
distributes to non-target tissues that may result in toxicity or adverse
reactions. Selective and targeted drug therapy could result in not just optimum
and more effective therapy but also a significant reduction in drug dose and
cost.
Targeted- or site-specific DDS refer to systems that place the drug at
or near the receptor site or site of action.
Site-targeted DDS can be classified into three
broad categories –
1. First-order targeting – refers to DDS that delivers the drug to the capillary bed or the
active site
2. Second-order targeting – refers to DDS that delivers the drug to a special cell type
such as the tumour cells and not to the normal cells
3. Third-order targeting – refers to DDS that delivers the
drug intracellularly.
Site-targeted DDSs have also been characterized as
–
Passive targeting – refers to natural or passive disposition of a drug carrier
based
on the physicochemical characteristics of the system in relation to the
body.
Active targeting – refers to alterations of the natural disposition of the drug carrier,
directing it to specific cells, tissues or organs; for e.g. use of ligands or
monoclonal antibodies which can target specific sites.
Drug targeting often requires carriers for selective
delivery and can serve following purposes –
·
Protect the drug from degradation
after administration;
·
Improve transport or delivery of
drug to cells;
·
Decrease clearance of drug; or
·
Combination of the above.
Carriers for drug targeting are of two types –
·
Carriers covalently bonded to
drug – where the drug release is required
for
pharmacological activity.
·
Carriers not covalently bonded to
drug – where simple uncoating of the drug
is
required for pharmacological activity; e.g. liposomes. The various
carriers used for drug targeting are –
a. Polymeric carriers
b. Albumin
c. Lipoproteins
d. Liposomes.
1. Polymeric Carrier Systems for Drug Targeting
The basic components of a
polymeric targeted DDS are –
1. A polymeric backbone which
is non-immunogenic and biodegradable that contains following three attachments;
2. A homing device, also
called as site-specific targeting moiety, which is capable of leading the
drug delivery system to the vicinity of a target tissue (or cell);
3. A solubiliser, which
enables the drug delivery system to be transported to and preferentially taken
up by the target tissue; and
4. A drug, which is covalently bonded to
the polymeric backbone, through a spacer, and contains a linkage that is
cleavable only by a specific enzyme(s) at the target tissue (fig. 14. 7).
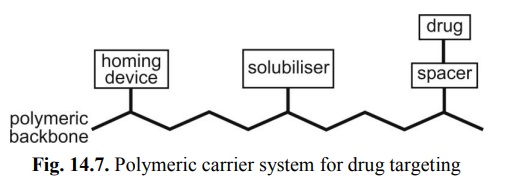
Fig. 14.7. Polymeric carrier system for drug
targeting
Polymers used for drug targeting include
polyethylenediamine, polylysine, chitosan, dextran and PEG for macromolecular
drugs such as gene therapy. The homing device is a monoclonal antibody, a
recognised sugar moiety or a small cell-specific ligand. At present, most
site-specific DDS are limited to parenteral administration and primarily
utilise soluble polymers.
Besides their use as regular carriers, polymers may
also be formulated as microparticles or nanoparticles, wherein the drug is
encapsulated in a biodegradable colloidal
polymer. The small size of nanospheres allows good tissue penetration while
providing protection or sustained release.
The disposition of micro- or nano-sphere depends
upon their size –
·
Particles > 12 μ are lodged in the capillary bed at the site of injection
·
Particles from 2 – 12 μ are retained in lung, liver or spleen
·
Particles < 0.5 μ (500 nm) deposit in spleen and bone marrow.
2. Albumin as Carrier for Drug Targeting
Although distribution of albumin is not site-specific,
it has been conjugated with drugs such as methotrexate to increase duration of
drug action and deliver drug to liver.
3. Lipoproteins as Carrier for Drug Targeting
Low-density lipoproteins enter cell by endocytosis
and thus have the potential for transporting drugs into the cell in which
lipoprotein-drug complex can be hydrolysed by lysosomal enzymes.
4. Liposomes as Carrier for Drug Targeting
Liposomes in the size range 0.5 – 100 have been used to reduce side effects and
efficacy of drugs such as doxorubicin, amphotericin B, etc. The
site-specificity to liposomes can be conferred by the type of lipid or by
inclusion of a targeting agent such as monoclonal antibody into the liposomal
bilayer.
Related Topics