Construction of Genomic Libraries
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Recombinant DNA Technology
Before we study how genomic libraries are made we first need to understand the differences between the genetic organization in eukaryotic and prokaryotic cells. Bacterial genes are uninterrupted sequences of nucleotides encoding the genetic information required for the synthesis of a protein.
CONSTRUCTION OF GENOMIC LIBRARIES
Before we study how genomic libraries are made we first need to understand the differences between the genetic organization in eukaryotic and prokaryotic cells. Bacterial genes are uninterrupted sequences of nucleotides encoding the genetic information required for the synthesis of a protein. These genes can sometimes be cotranscribed with adjacent genes of related function into the same mRNA molecule. This set of cotranscribed genes is called an operon (Figure 25.3). The mRNA in bacteria does not generally need to be processed before translation, and transcription and translation occur simultaneously.
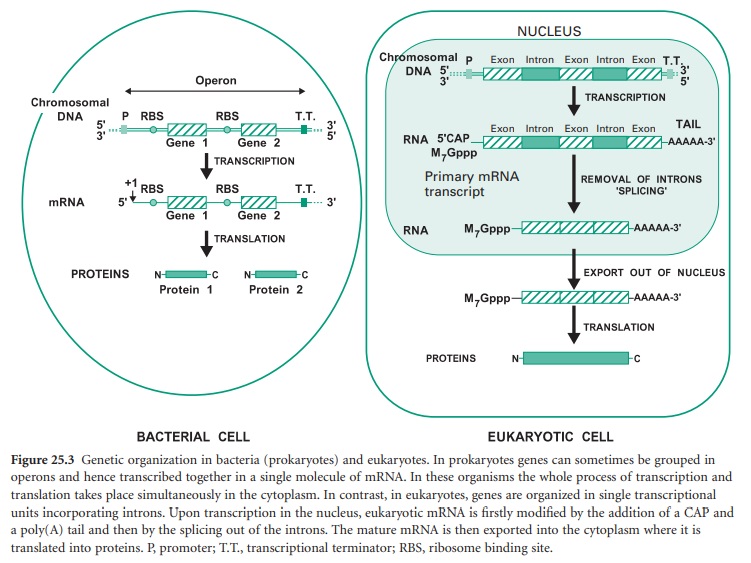
In contrast, genes from eukaryotic cells contain non-coding sequences called introns and coding sequences called exons. The former are removed after transcription by a process called ‘splicing’ that occurs in the nucleus of the cell. In addition the mRNA is subjected to further processing involving the addition of a methylated guanine (M7Gppp) called CAP on its 5′ end, required for translation, and a polyadenine (poly(A))tail on its 3′ end (Figure 25.3). Mature mRNA is then exported from the nucleus into the cytoplasm, where it is translated into proteins. Eukaryotic genes appear to be transcribed individually, as operons have not been described in eukaryotes.
To enable the cloning and isolation of a specific gene(s) from a cell several steps are required. The first consists of choosing the source of genetic material which, in prokaryotic cells, is normally the chromosomal DNA. In contrast, in eukaryotic cells this is more often the mature mRNA, as it is not interrupted by introns and consequently codes for complete, active proteins. The second step consists of the preparation of the purified DNA or RNA for cloning. This step is more straightforward when using prokaryotic DNA than eukaryotic RNA. The result will be the construction of a collection of cloned DNA fragments propagated in bacteria that is called the genomic library. This library should ideally contain representatives of every sequence in the chromosome of a prokaryotic cell and every expressed gene in the case of a eukaryotic cell. The final step consists of the screening of the recombinant clones to identify the required gene(s).
i) Prokaryotic gene libraries: shotgun cloning
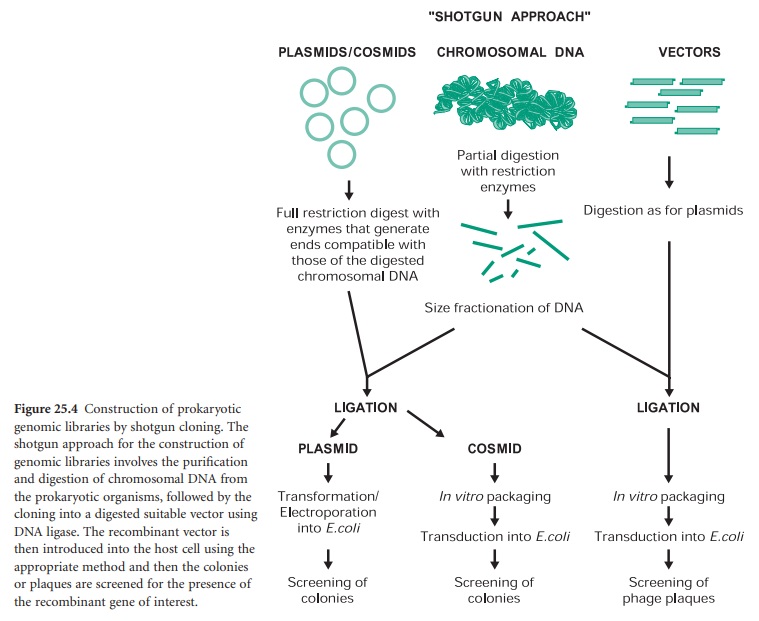
The construction of a prokaryotic gene library can be achieved by a technique called ‘shotgun cloning’ (Figure 25.4). This involves the purification and partial digestion of the genomic (chromosomal) DNA from a prokaryotic organism with restriction endonucleases to produce a random mixture of fragments of different sizes. Chromosomal DNA can also be mechanically sheared and in this case the extremities must be repaired and made blunt with DNA polymerase in the presence of deoxynucleotides (dNTPs). These fragments are then fractionated into different sizes and ligated into a cloning vector appropriately digested. The recombinant vectors are then transformed, in the case of plasmids, or transfected, in the case of bacteriophages and cosmids, into the host cell of choice. The resulting genomic library can then be screened for the presence of the recombinant gene of interest by a number of methods.
ii) Eukaryotic cDNA gene libraries
The shotgun approach cannot be applied for the construction of eukaryotic gene libraries because of the presence of introns in the DNA, which prevents the direct cloning of functional genes from digested chromosomal DNA. Instead, mature mRNA from the cytoplasm of cells expressing the desired gene is used as the source of genetic material. For example, to make a genomic library containing the insulin gene, RNA from pancreatic cells expressing this gene have to be isolated. Remember that cells show distinct differentiation in different tissues, and express only a small percentage of the whole genome according to their role in the tissue of which they form part. Consequently, it is not possible to purify RNA coding for insulin from, for example, cells of the pituitary gland. Therefore, the cells expressing the gene of interest have to be isolated first, and then their mRNA purified. As mentioned earlier, virtually every eukaryotic mRNA has a poly(A) tail on its 3′ end. This provides a convenient way to isolate mature mRNA from total cellular RNA, most of which (98%) is ribosomal RNA (rRNA) and transfer RNA (tRNA). The total RNA purified from a cell can be passed through an affinity column packed with cellulose linked to deoxythymidine oligonucleotides (oligo(dT)). As the total RNA passes through the column, only the mRNA molecules which have poly(A) tails will bind to the oligo(dT) while the rest will flow through the column to be discarded. The purified mRNA then has to be converted into ds cDNA (complementary DNA) to enable its cloning into a suitable vector.
· Synthesis and cloning of cDNA
There are generally two main strategies used for the synthesis of cDNA from mRNA: replacement synthesis and primer adaptor synthesis. For both strategies the first strand cDNA synthesis is based on the priming of the mRNA with an oligo-dT which anneals to the poly(A) tail of the mRNA molecule and, consequently, with the action of the enzyme reverse transcriptase and in the presence of dNTPs, the synthesis of the first cDNA strand takes place. This results in the formation of a mRNA/cDNA heteroduplex hybrid. The second stage is different for the two strategies mentioned. The most commonly used is the replacement synthesis, which is based on the use of ribonuclease H (RNaseH), an enzyme that cleaves the RNA moiety of RNA/DNA hybrids and has 5′ to 3′ and 3′ to 5′ direction exonuclease activities. This results in partial digestion of the RNA in both directions. The resulting RNA fragments can serve as primers for DNA synthesis using DNA polymerase I. This enzyme, with its 5′ to 3′ direction exonuclease and polymerase activities will fill the nicks and effectively remove the RNA primers. The cDNA fragments synthesized will be joined using DNA ligase. This method causes the loss of some nucleotides at the 5′ end of the mRNA, including the CAP region.
The primer adaptor method for the synthesis of the second strand of cDNA starts with the removal of the RNA strand from the mRNA/cDNA hybrid, by treatment with alkali. This is followed by the addition of a poly(C) tail to the 3′ end of the DNA strand using an enzyme called terminal transferase. This enables the hybridization of a complementary poly(G) primer that will be the starting point for the synthesis of the second cDNA strand by the DNA polymerase (Figure 25.5). This method, in contrast to the replacement synthesis, generates cDNA molecules with a complete 5′ CAP region. However, it requires more steps and the terminal transferase step is difficult to control.
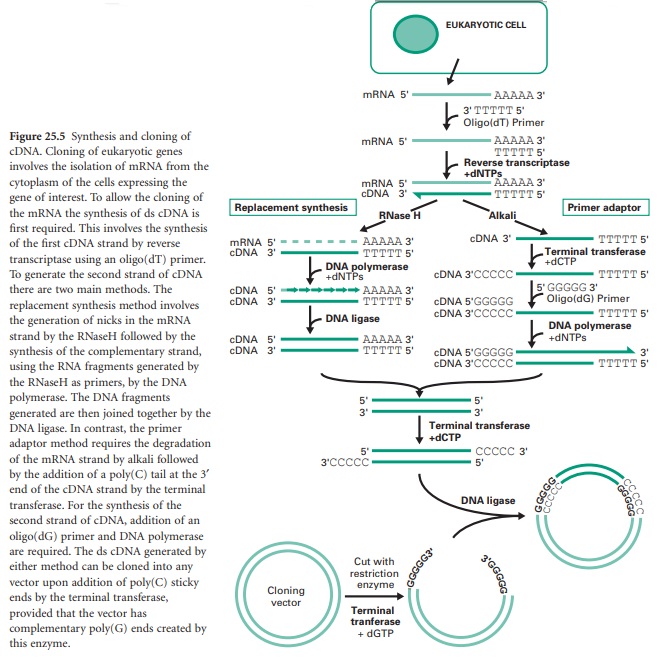
Finally, the cloning of the resulting cDNA is aided by the addition of a poly(C) tail at the 3′ ends of the cDNA fragments using terminal transferase and the ligation of these to a linearized vector containing a complementary poly(G) tail also generated by the terminal transferase.
iii) Comparison
between libraries
There are a number
of points to take into
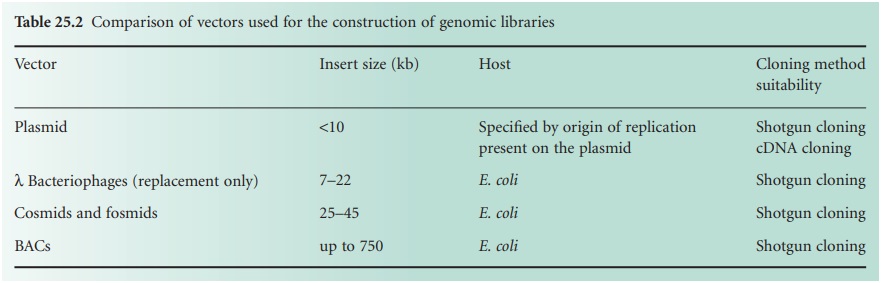
consideration when choosing a strategy to generate a genomic library. The larger the insert
size, the lower
the number of clones
required to have full
representation of an entire genome. To reduce
the number of recombinant clones to be screened before the gene of interest
is identified, cosmids, λ bacteriophages or BACs should
be used as they can take
larger
DNA inserts. Furthermore, if we want
to isolate a large DNA fragment containing, for example, all the
genes required for the biosynthesis of an antibiotic, plasmid libraries might not be the right choice. In contrast, if we are trying
to isolate a single gene and the extent of the screening is not a problem, plasmid
libraries are ideal
as they reduce
the subcloning steps
required to single out the gene of interest. Table 25.2 shows a comparison between the
insert sizes taken
by different vectors, their hosts and the genomic
libraries for which
they can be suitable.
Related Topics
