Control of Microbial Contamination During Manufacture: General Aspects
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Principles Of Good Manufacturing Practice
A pharmaceutical product may become contaminated by a number of means and at several points during manufacture. There are several ways in which this risk can be minimized. Any such measures require an understanding of the risks involved.
CONTROL OF MICROBIAL CONTAMINATION DURING MANUFACTURE: GENERAL ASPECTS
A pharmaceutical product may become contaminated by a number of means
and at several points during manufacture. There are several ways in which this
risk can be minimized. Any such measures require an understanding of the risks
involved.
A) Risk Assessment
GMP is informed by past mistakes and case studies have been valuable
(Friedman, 2004). However a proactive approach is required. Nowadays a
manufacturer is expected to demonstrate to the regulatory authorities that an
extensive risk assessment has been carried out. Risk analysis must comply with
ICH 9Q (EMEA, 2006) and is underpinned by a sound understanding of the process
and of the microbial ecology of the environment and ingredients.
Several methods are employed (see EMEA, 2006; Kirupakar, 2007),
including hazard analysis critical control points (HACCP), failure mode and
effects analysis (FMEA), fault tree analysis (FTA), risk ranking and filtering
(RRF) and hazard operability analysis (HAZOP). Only HACCP and FMEA are discussed
here.
i) Hazard analysis critical control
points (HACCP)
HACCP has been widely used in the food industry and is becoming more
commonly used in the pharmaceutical industry (McCullogh, 2007; Sharp, 2000;
WHO, 2003; Whyte, 2010). The original HACCP had seven steps:
1.
Conduct a hazard analysis and identify
preventive measures for each step of the process
2.
Determine the critical control points.
3.
Establish critical limits.
4.
Establish a system to monitor the
critical control points.
5.
Establish the corrective action to be
taken when monitoring indicates that the critical control points are not in a
state of control.
6.
Establish a system to verify that the
HACCP procedure is working effectively.
7.
Establish a record-keeping system.
However, HACCP has been modified so that it can be applied quantitatively
not only to microbiology but also to pyrogens and particles (Tidswell, 2004;
Whyte, 2010).
ii) Failure mode and effects
analysis (FMEA)
FMEA was first used in the engineering industry (Stamatis, 2003). It
involves breaking the process down into many discrete steps. For each step
scales are set for severity, occurrence and detection. The scores are
multiplied and compared to an informed score at which risk becomes
unacceptable.
B) Environmental Cleanliness And Hygiene
Microorganisms may be transferred to a product from working surfaces,
fixtures and equipment. Pooled stagnant water is a frequent source of
contamination. Thus it is essential that all working areas are kept clean, dry
and tidy. Any cracks where microorganisms may accumulate must be eliminated.
All walls, floors and ceilings should be easy to clean. This entails impervious
and washable surfaces, free from open joints or ledges. Coving should be used
at junctions between walls and floors or ceilings. All services such as pipes,
light fittings and ventilation points should be sited so that inaccessible
recesses are avoided. A rigorous disinfection policy must be in place. All
equipment must be easy to dismantle and clean and should be inspected for
cleanliness before use.
Fall-out of dust-and droplet-borne microorganisms from the atmosphere is
an obvious route for contamination. ‘Clean’ air is therefore a prerequisite
during manufacturing processes and the spread of dust during manufacture or packaging
must be avoided. Microorganisms may thrive in certain liquid preparations and
creams and ointments. The manufacture of such products should, as far as possible,
be in a closed system; this serves a dual purpose as it also prevents evaporative
loss.
Personnel are another source of potential contamination. High standards
of personal hygiene are essential. Operatives should be free from communicable
disease and open lesions on exposed body surfaces. To ensure high standards of
personal cleanliness, adequate hand-washing and hand-disinfecting facilities
and protective garments, including headgear and gloves, must be provided. Staff
should be trained in the principles of GMP and in the practice (and theory) of
the tasks assigned. Staff employed in the manufacture of sterile products
should also receive training in basic microbiology.
C) Quality Of Starting Materials
Raw materials account for a high proportion of the microorganisms
introduced during the manufacture of pharmaceuticals, and the selection of
materials of good microbiological quality aids in the control of contamination
levels in both products and the environment. It is, however, common to have to
accept raw materials which have some non-pathogenic microorganisms present and
this must be considered during risk assessment. Whatever the means of
prevention of growth or survival by chemical or in-process treatment, it should
be regarded as critical and controlled accordingly (Sharp 2000).
Untreated raw materials that are
derived from a natural source usually support an extensive and varied
microflora. Products from animal sources such as gelatin, desiccated thyroid,
pancreas and cochineal may be contaminated with animal-borne pathogens. For
this reason some statutory bodies such as the British Pharmacopoeia require
freedom of such materials from Escherichia coli and Salmonella spp. At a stated level before they can
be used in the preparation of pharmaceutical products. The microflora of
materials of plant origin such as gum acacia and tragacanth, agar, powdered
rhubarb and starches may arise from those indigenous to plants and may include
bacteria such as Erwinia spp., Pseudomonas spp., Lactobacillus spp., Bacillus spp., streptococci, moulds such as Cladosporium spp., Alternaria spp.
And Fusarium spp. And non-mycelated yeasts, or those
introduced during cultivation. For example, the use of untreated sewage as a
fertilizer may result in animal-borne pathogens such as Salmonella spp. Being present. Some refining
processes modify the microflora of raw materials; for example, drying may
concentrate the level of spore-forming bacteria and some solubilizing processes
may introduce waterborne bacteria such as Escherichia coli.
Synthetic raw materials are usually free from all but incidental microbial
contamination.
The storage condition of raw materials,
particularly hygroscopic substances, is important, and as a minimum water
activity (Aw) of 0.70 is
required for osmophilic yeasts, 0.80 for most spoilage moulds and 0.91 for most
spoilage bacteria, precautions should be taken to ensure that dry materials are
held below these levels. Some packaging used for raw materials,
such as unlined paper sacks, may absorb moisture and may itself be subject to
microbial deterioration and so contaminate the contents; for this reason
polythene-lined sacks are preferable. Some liquid or semisolid raw materials
contain preservatives, but others such as syrups depend upon osmotic pressure
to prevent the growth of osmophiles, which are often present. With this type of
material it is important that they are held at a constant temperature, as any
variation may result in evaporation of some of the water content followed by
condensation and dilution of the surface layers to give an Aw value which
may permit the growth of osmophiles and spoil the syrup.
The use of natural products with a high
non-pathogenic microbial count is possible if a sterilization stage is included
either before or during the manufacturing process. Such sterilization
procedures may include heat treatment, filtration,
irradiation, recrystallization from a bactericidal solvent such as an alcohol,
or for dry products, where compatible, ethylene oxide gas. If the raw material
is only a minor constituent and the final product is adequately preserved
either by low Aw, chemically or
by virtue of its pH, sugar or alcohol content, an in-process sterilization
stage may not be necessary. If, however, the product is intended for parenteral
or ophthalmic use a sterilization stage is essential.
The handling of contaminated raw materials as described previously may
increase the airborne contamination level, and if there is a central dispensing
area precautions may be necessary to prevent airborne cross-contamination, as
well as that from contaminated measuring and weighing equipment. This presents
a risk for all materials but in particular those stored in the liquid state
where contamination may result in the bulk being spoiled.
D) Water
Many grades of water are used in
pharmaceutical manufacturing (Table 23.1).
Water for manufacturing may be potable mains water, water purified by ion
exchange, reverse osmosis or distillation, or water for injection purposes
(EMEA, 2002).
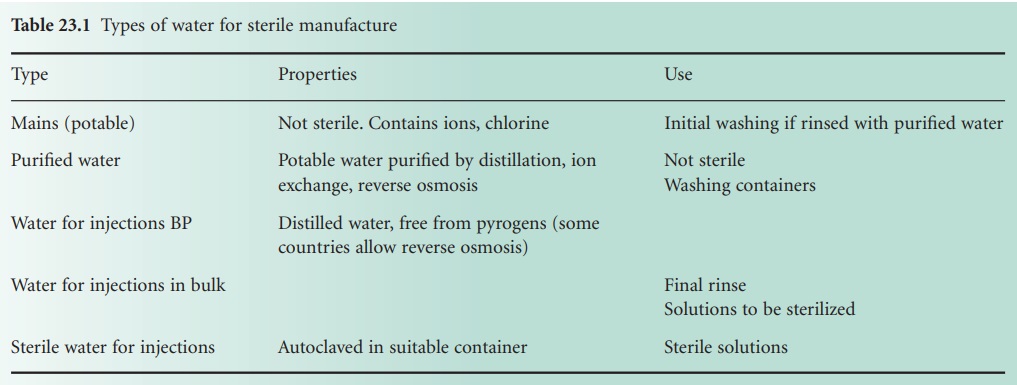
Most types of water are derived from municipal supplies. Such water is
treated, sometimes by filtration, and always by chemicals, usually chlorine, to
render it free from coliforms. This water is, however, not sterile. Its
microbial and chemical content varies from region to region and the microbial
count can increase on storage.
Water used for parenteral products,
known as Water for Injections or Water For Injection (WFI), must be virtually
apyrogenic. The British Pharmacopoeia (British
Pharmacopoeia Commission, 2010) and the US Pharmacopeia (2009a)
specify an endotoxin level of no more than 0.25 IU/ml for WFI. In Europe such
water is usually produced in a still specially designed to prevent pyrogens
from being mechanically carried over into the distillate. In other countries
reverse osmosis may also be used (US Pharmacopoeia, 2009a), but in Europe
reverse osmosis is not approved (EMEA, 2002). WFI can be used immediately for
the preparation of injections, provided it is sterilized within 4 hours of
water collection. Alternatively, the water can be kept for longer periods at a
temperature above 65°C (typically 80°C) to prevent bacterial growth with
consequent pyrogen production. Ultraviolet radiation may be useful for treating
WFI in order to reduce the bacterial count, but this must not be regarded as a
sterilization process . A more detailed account of water for pharmaceutical use
may be found in EMEA (2002) and US Pharmacopeia (2009b).
E) Process Design
The manufacturing process must be fully defined and capable of
providing, with the facilities available, a product that is microbiologically
acceptable and conforms to specifications. The process must be fully validated
before starting to ensure that it is suitable for routine production
operations. Processes and procedures must also be subject to frequent
reappraisal and should be re-evaluated when any significant changes are made in
the equipment or materials used.
F) Quality Control And Documentation
The lower the microbiological count of the starting materials, the more
readily the quality of the product can be controlled. Microbiological standards
should be set for all raw materials as well as microbial limits for in-process
samples and the final product. Microbiological quality assurance also covers the
validation of cleaning and dis-infectant solutions and the monitoring of the
production environment by microbial counts. This monitoring should be carried
out while normal production operations are in progress. In addition, sterile
manufacture requires extra safeguards. Operators must be adequately trained and
their aseptic technique monitored both by observation and microbiological
testing. Air filter and sterilizer efficiency must also be evaluated , whilst
sterility testing and, where necessary
testing for pyrogens , are the final tests on the finished product.
Documentation is a vital part of quality assurance. Details of starting
materials, packaging materials, and intermediate, bulk and finished products
should be recorded so that the history of each batch may be traced.
Distribution records must be kept. This information is of paramount importance
in the event that a defective batch has to be recalled.
G) Packaging, Storage And Transport
Packaging serves a number of functions; it keeps the contents in, it
should keep contaminants out and is labelled to permit identification of its
contents. The product is contained within primary packaging. In industry these
packages are then placed inside secondary packaging for storage and transport.
This secondary packaging may take the form of cartons, boxes, trays or shrink
wrapping.
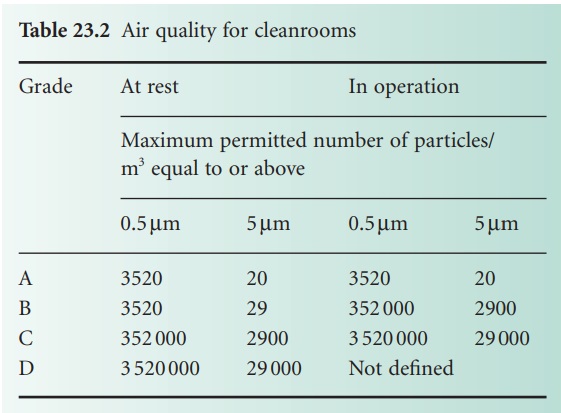
Consideration must be given to both the
fabric of the packaging and its cleaning, and to the actual process of
packaging. Where terminal sterilization is carried out, the packaging must be
suitable for the process. Packaging of aseptically processed products into a
sterile container must be carried out in a grade A environment (Table 23.2).
Packaging material
has a dual role and acts both to
contain the product and to prevent the entry of microorganisms or moisture
which may result
in spoilage, and it
is therefore important that the source
of contamination is not the
packaging itself. The
microflora of a packaging
material is dependent upon both its composition and storage conditions. This, and a consideration of the type of pharmaceutical product to be
packed, determines whether a sterilization treatment is required.
Glass containers are sterile on leaving the furnace, but are often stored
in dusty conditions and packed for transport in cardboard boxes. As a result they may
contain mould
spores of Cladosporium spp., Penicillium spp., Aspergillus
spp. and bacteria such as Bacillus spp.
and Micrococcus spp. which
originate from the cardboard,
although it can be treated
to remove these contaminants.
It is commonplace either
to air-blow or wash glass containers to remove
any glass spicules
or dust which may be present,
and it is often advantageous to include a disinfection stage if the
product is a liquid or semisolid
preparation. Plastic
bottles that are either blowor injection-moulded have a very
low microbial count
and may not require disinfection. They may, however,
become contaminated with
mould spores if they are transported
in a non-sanitary packaging material
such as unlined cardboard. Packaging materials
that have a smooth, impervious
surface, free from crevices or interstices,
e.g. cellulose acetate, polyethylene, polypropylene, polyvinyl chloride (PVC),
and metal foils
and laminates, all have a low surface microbial count.
Closure
liners of pulpboard or cork, unless specially treated with a preservative, foil or wax coating,
are often a source of mould contamination for liquid or semisolid products. A closure
with a plastic
flowed-in liner is less prone to introduce or support
microbial growth than one stuck in with an adhesive, particularly if the latter
is based on a natural product such as casein. Closures
can be sterilized
by either formaldehyde or ethylene
oxide gas if required. In the case of injectables and ophthalmic preparations which are manufactured aseptically but do not receive
a sterilization
treatment in their
final container the
packaging has to be sterilized (Figure
23.2b). Dry heat
at 170 °C is often used for vials and ampoules.
Containers and closures
may also be sterilized by moist heat, chemicals and irradiation,
but consideration of the destruction or removal of bacterial pyrogens may be necessary.Regardless of the type of sterilization, the process must be validated and critical control
points or other risk assessment parameters must be established.
Related Topics
