Dissolution
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Biopharmaceutical considerations
For most drugs, the rate at which the solid drug dissolves in a solvent (dis-solution) is often the rate-limiting step in the drug’s bioavailability.
Dissolution
For
most drugs, the rate at which the solid drug dissolves in a solvent
(dis-solution) is often the rate-limiting step in the drug’s bioavailability.
Noyes–Whitney equation
Noyes–Whitney
equation correlates the dissolution rate of a drug with the particle surface
area (S), the thickness of the
unstirred solvent layer on the particle surface (h), diffusion coefficient of the drug (D), and the concentra-tion gradient, that is, the difference in the
concentration of drug solution at the particle surface (Cs) and the bulk solution (C).
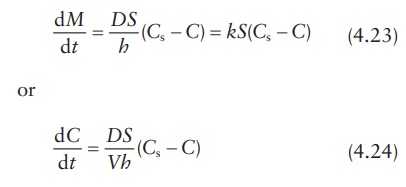
where:
dM /dt
is the mass rate of dissolution (mass of the drug dissolved per unit time,
e.g., in mg/min)
D is the diffusion
coefficient of solute in solution, in cm2/s
S is the surface area
of the exposed solid, in cm2
k is the dissolution
rate constant (k = D /h, in cm/s)
h is the thickness of
the unstirred layer at the solid surface, in cm
Cs is the drug solubility at the particle surface, in g/mL
C is the drug
concentration in bulk solution at time t,
in g/mL
The
quantity, dC/dt, represents the change in drug concentration in the bulk solution
per unit time, or the dissolution rate, and V
is the volume of solution (mL). Thus, C
= M /V.
Under
sink conditions, C << Cs. Therefore, the
Noyes–Whitney equation can be simplified as:
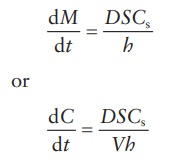
Calculation example
Knowledge
of dissolution rate constant, k,
allows simulation of the rate of drug dissolution by using different quantities
of drug substance, changes in the particle size and surface area of the drug,
and dissolution condi-tions, such as volume. A simulation of these results can
assist in dissolution method development by minimizing the number of
experiments needed under different conditions. Dissolution rate constant can be
calculated using dissolution data collected from a well-defined system.
For
example, a preparation of drug particles weighing 550 mg and having a total
surface area of 0.28 × 104 cm2 was allowed to dissolve in
500 mL of water at 37°C. Assuming that analysis of bulk dissolution sample showed
that 262 g had dissolved after 10 min, if the saturation solubility of the drug
in water is 1.5 mg/mL at 37°C, k can
be calculated as follows:
According
to the Noyes–Whitney equation,

The
dissolution rate constant is related to the diffusion constant of the drug
through the solvent (D) and the
diffusion layer thickness (h):
k = D/h (4.25)
Therefore,
if the diffusion layer’s thickness could be estimated, the diffu-sion
coefficient of the drug can be calculated. Thus, if the diffusion layer’s
thickness were 5 × 10–3 cm, the diffusion coefficient (D) would be given by:
0.
0201 cm/min = D / (5 × 10 −3 cm)
or
D = 0. 0201 cm/min × 5 × 10 −3 cm = 1.01×10−4 cm2 / min
Factors influencing dissolution rate
The
main biopharmaceutical and physiological factors that influence the dissolution
rate of a drug can be summarized as follows:
1. Drug solubility: The greater the
drug solubility, the greater the drug’s dissolution
rate. This is evident in the Noyes–Whitney equation. The solubility and
dissolution rates of acidic drugs are low in acidic gastric fluids, whereas the
solubility and dissolution rates of basic drugs are high. Similarly, the
solubility and dissolution rates of basic drugs are low in basic intestinal
fluids, whereas those of acidic drugs is high.
2. Viscosity (of the dissolving
medium): The greater the viscosity of the
dissolving liquid, the lower the diffusion coefficient of the drug and
hence the lower the dissolution rate. Viscosity of the dissolving bulk medium
and/or the unstirred layer on the surface of the dissolving formulation can be
affected by the presence of hydrophilic polymers in the formulation, which
dissolve to form a viscous solution. In
vivo, the viscosity may be affected by the food intake.
3. Diffusion layer’s
thickness:
The greater the diffusion layer’s thickness,
the slower the dissolution rate. The thickness of the diffusion layer is
influenced by the degree of agitation of the dissolving medium, both in vitro and in vivo. Hence, an increase in gastric and/or intestinal motility may increase the dissolution
rate of poorly soluble drugs. For example, food and certain drugs can influence
gastrointestinal (GI) motility.
4. Sink conditions: Removal rate of
dissolved drugs by absorption through
the GI mucosa and the GI fluid volume affect drug concen-tration in the GI
tract.
5. pH (of the dissolving
medium): The drug dissolution rate is deter-mined by the drug solubility in the
diffusion layer surrounding each dissolving drug particle. The pH of the
diffusion layer has a significant effect on the solubility of a weak
electrolyte drug and its subsequent dissolution rate. The dissolution rate of a
weakly acidic drug in GI fluid (pH 1–3) is relatively low because of its low
solubility in the diffusion layer. If the pH in the diffusion layer could be
increased, the solubility exhibited by the weak acidic drug in this layer (and
hence the dissolution rate of the drug in GI fluids) could be increased. The
potassium and sodium salt forms of the weakly acidic drug have a relatively
high solubility at the elevated microenvironmental pH in the diffusion layer
due to the strong counterion bases, KOH and NaOH, respectively. Thus, the
dissolution of the drug particles takes place at a faster rate.
6. Particle size and
surface area:
An increase in the specific surface area (surface
area per unit mass) of a drug in contact with GI fluids would increase its
dissolution rate. Generally, the smaller a drug’s particle size, the greater
its specific surface area and the higher the dissolution rate. However,
particle size reduction may not always be helpful in increasing the dissolution
rate of a drug and hence its oral bioavail-ability. For example:
·
Porosity of drug particles
plays a significant role. Thus, smaller particles
with lower porosity may have lower surface area com-pared with larger particles
with greater porosity. The dissolution rate depends on the effective surface area, which includes the influence of particle
porosity.
·
In some cases, particle size reduction may cause particle aggre-gation, thus reducing the
effective surface area. To prevent the formation
of aggregates, small drug particles are often dispersed in polyethylene glycol
(PEG), polyvinylpyrrolidone (PVP), dextrose, or surfactants such as
polysobrates. For example, micronized griseofulvin is dispersed in PEG 4000.
·
In addition, certain drugs such as penicillin G and
erythromycin are unstable in gastric
fluids and do not readily dissolve in them. For such drugs, particle size
reduction may increase not only the rate of drug dissolution in gastric fluids
but also the extent of drug degradation.
7. Crystalline
structure:
Amorphous (noncrystalline) forms of a drug
may have faster dissolution rate compared with the crystalline forms. Some
drugs exist in a number of crystal forms or polymorphs. These different forms
may have significantly different drug solubility and dissolution rates.
a. Dissolution rate of a drug from a crystal form is a
balance between the energy required to break the intermolecular bonds in the
crys-tal and the energy released on the formation of the drug–solvent
intermolecular bonds. Thus, stronger crystals may have lower intrinsic
dissolution rate.
b. Intrinsic
dissolution rate reflects the dissolution rate of a drug crystal or powder normalized for its surface area. It is expressed
in terms of mass per unit time per unit surface area. Drug forms that have
higher intrinsic dissolution rate are expected to have higher dissolution
rates.
c. The greater strength of a crystalline polymorph,
sometimes evi-dent by its high melting
point and sometimes by the rank order, correlates with its lower intrinsic
dissolution rate.
d. Similarly, amorphous solids, which lack a long-range
order that defines crystalline structure, tend to have higher intrinsic
dissolu-tion rates.
8. Temperature: An increase in
temperature leads to greater solubil-ity of a solid, with positive heat of the solution. Heat of solution indicates release
of heat on dissolving. Positive heat of solution is indicative of a greater
strength of solute–solvent bonds formed (which release energy) compared with
the solute–solute bonds bro-ken (which take energy). The solid will therefore
dissolve at a more rapid rate if the system is heated. Therefore, in vitro dissolution studies are carried
out at 37°C to simulate body temperature and in vivo dissolution condition.
9. Surfactants: Surface-active
agents increase the dissolution rate by (a)
lowering the interfacial tension,
which lowers the contact angle of the
solvent on the solid surface and increases wetting of the drug particle and
penetration of the solvent inside the dosage form, and (b) increas-ing the
saturation solubility of the drug in the dissolution medium. Surfactants such
as sodium lauryl sulfate (SLS) and Triton X-100 are frequently used to achieve
sink conditions and rapid dissolution during in vitro dissolution method development.
Related Topics