Injections
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Sterile Pharmaceutical Products
Injections may be aqueous solutions, oily solutions (because of poor aqueous solubility or the necessity for a prolongation of drug activity), aqueous suspensions or oily suspensions.
INJECTIONS - TYPES OF STERILE PRODUCT
Injections may be aqueous solutions, oily solutions (because of poor
aqueous solubility or the necessity for a prolongation of drug activity),
aqueous suspensions or oily suspensions. They may be aseptically produced or
terminally sterilized in their final containers. Those drugs that are unstable
in solution may be presented as a freeze-dried (lyophilized) powder. The choice
of final packaging should not determine the method of sterilization.
A) Formulation Philosophy
An injection must be manufactured under
conditions that result in a product containing the minimum possible levels of
particles and pyrogenic substances. Its formulation and packaging must maintain
physical and chemical stability throughout the production processes, the
intended shelf life and during administration. To achieve this, excipients such
as buffers and antioxidants may be required to ensure chemical stability, and
solubilizers, such as propylene glycol or polysorbates, may be necessary for drugs
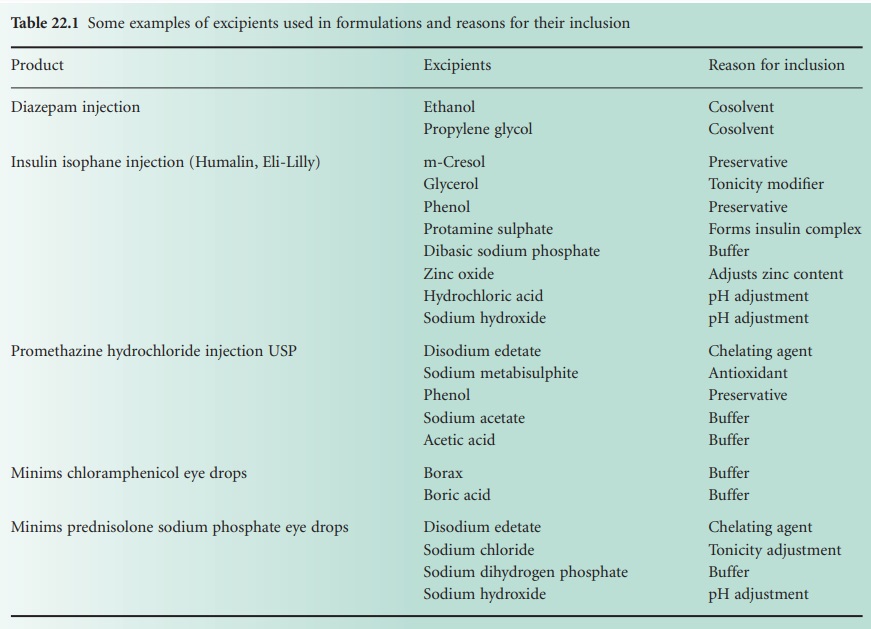
with poor aqueous solubility to maintain the drug in solution. Table 22.1 lists
some chemical constituents of common injections and ophthalmic preparations.
Many injections are formulated as aqueous solutions, with Water for
Injections as the vehicle. Their formulation depends on several factors
including the aqueous solubility of the active ingredient, the dose, its
thermal stability, the route of administration, and whether the product is to
be offered as a multiple-dose product (i.e. with doses removed on different
occasions) or as a single-dose form (as the term suggests, only one dose per
container). Most injections are prepared in single-dose form but this is
mandatory for certain routes, e.g. spinal injections where the intrathecal route
is used, and large-volume intravenous infusions. Multiple-dose injections may
require the inclusion of a suitable preservative to prevent contamination following
the removal of each dose. Injections used for several routes, including the
intrathecal and intracardiac routes, must not contain a preservative because of
potential long-term damage to the patient. A review of the preservatives used
in parenteral products has been given by Meyer & Shi (2009).
Some types of injections must be isotonic with blood serum. This applies
particularly to large-volume intravenous infusions if at all possible;
hypotonic solutions may cause lysis of red blood corpuscles and thus must not
be used for this purpose. Conversely, hypertonic solutions can be employed;
these induce shrinkage, but not lysis, of red cells, which recover their shape
later. Intraspinal injections must also be isotonic to reduce pain at the site
of injection; so should intramuscular and subcutaneous injections. Adjustment
to isotonicity can be determined from either the depression of freezing point
or from sodium chloride equivalents. The depression of the freezing point
depends on the number of dissolved particles (molecules or ions) present in a
solution. The equation:
![]()
where W is the
percentage (w/v) of adjusting substance, a is the
freezing point of unadjusted solution and b is the
depression of the freezing point of water induced by 1% w/v of adjusting
substance, allows the determination of how much adjusting substance is required
to produce isotonicity with blood plasma.
Alternatively the sodium chloride
equivalent, which is produced by dividing the value for the depression of
freezing point produced by a solution of the substance by the corresponding
value of a solution of sodium chloride of the same strength, may be used.
Fuller details of each method may be found in the Pharmaceutical Codex (1994).
B) Intravenous Infusions
Intravenous infusions consist of
large-volume injections or drips (500 ml or more) that are infused at various
rates (50–500 ml/h) into the venous system. They are generally sterilized in an
autoclave. Examples include isotonic solutions of sodium chloride or glucose
that are used to maintain fluid and electrolyte balance, for replacement of
extracellular body fluids (e.g. after surgery or prolonged periods of fluid
loss), as a supplementary energy source (1 L of 5% w/v glucose yields 714 kJ of
energy) or as a vehicle for drugs. Other important examples are blood products,
which are collected and processed in sterile containers, and plasma
substitutes, e.g. dextrans and degraded gelatin. Dextrans are glucose polymers
in which the glucose monomers are joined by 1–6-α links; they are produced by
certain bacteria of the genus Leuconostoc,
e.g. Leuconostoc mesenteroides.
i) Intravenous additives
A common hospital practice is to add drugs to infusions immediately
before administration. Regularly used additives include potassium chloride,
lidocaine (lignocaine), heparin, certain vitamins and antibiotics. Potentially
this can be a hazardous practice. For instance, the drug may precipitate in the
infusion fluid because of the pH (e.g. amphotericin) or the presence of calcium
salts (e.g. thiopentone); the drug may degrade rapidly (e.g. ampicillin in 5%
w/v glucose); multiple additions may lead to precipitation of one or both of
the drugs or to accelerated degradation; and finally, drug loss may occur
because of sorption by the container. For instance, insulin is adsorbed by
glass or by polyvinyl chloride (PVC); glyceryl trinitrite and diazepam are
absorbed by PVC. Apart from these problems, if the addition is not carried out
under strict aseptic conditions the fluid can become contaminated with
microorganisms during the procedure. Thus any addition should be made in a
laminar-flow workstation or isolator, and the fluid should, ideally, be
administered within 24 hours of preparation.
Another approach to the problem of providing an intravenous drug
additive service is to add the drug to a small volume (50–100 ml) infusion in a
collapsible plastic container and store the preparation at −20°C in a freezer.
The infusion can be removed when required and thawed rapidly in a microwave
oven. Many antibiotics are stable for several months when stored in minibags at
−20°C and are unaffected by the thawing process. Other antibiotics, e.g.
ampicillin, degrade even when frozen.
ii) Total parenteral
nutrition
Total parenteral nutrition (TPN) is the use of mixtures of amino acids,
vitamins, electrolytes, trace elements and an energy source (glucose and fat)
in the long-term feeding of patients who are unconscious or unable to take
food. All or most of the ingredients to feed a patient for 1 day are combined
aseptically in one large (3 L capacity) collapsible plastic bag, the contents
of which are infused over a 12–24 hour period. Transfer of amino acid, glucose
and electrolyte infusions, and the addition of vitamins and trace elements must
be carried out with great care under aseptic conditions to avoid microbial
contamination. These solutions often provide good growth conditions for
bacteria and moulds. Fats are administered as oil-in-water emulsions comprising
small droplets of a suitable vegetable oil (e.g. soyabean) emulsified with egg
lecithin and sterilized by autoclaving. In many cases, the fat emulsion is
added to the 3 L bag. Thus TPN fluids are complex mixtures and a multitude of
potential interactions, both chemical and physicochemical, may occur between
their individual components resulting in decomposition, creaming, precipitation
or even the formation of toxic by-products. Trace elements, calcium, vitamins
and lipids are particularly prone to affecting the stability.
Although many vitamins may be administered as a single dose at various
time intervals, many of the patient’s requirements will be found in what is
basically an emulsion formulation, prepared aseptically and thus with no
terminal sterilization. The product usually contains both essential and
non-essential amino acids rather than fully formed protein, and energy is
provided at a ratio of 0.6–1.1 MJ per gram of protein nitrogen. A mixture of
carbohydrate (glucose) and fat (as an emulsion) provides the energy and electrolytes,
trace elements and vitamins are included as required. Thus the TPN fluid is
prepared to suit the individual patient’s needs. The fact that the product
contains so many ingredients makes TPN fluids extremely difficult to prepare,
and once vitamins are added, their chemical instability reduces the shelf life.
During preparation, TPN fluids are compounded from individual solutions or
emulsions. Generally, the bulk of the final volume is derived from glucose
solutions, amino acid solutions and fat emulsions; small-volume solutions are
added to these before filling. During compounding, electrolytes are added to
the amino acid solutions and phosphate salts to the dextrose (glucose)
solutions, which are then mixed and the lipid emulsions added. This order of
mixing is adopted because the pH of glucose solutions decreases due to
degradation during their sterilization and addition of emulsions to this low pH
solution might cause emulsion instability. The mixing of the amino acids with
the glucose solutions provides a vehicle with some degree of buffering
capacity. Calcium might precipitate as the phosphate if its salts were to be
added directly to the phospholipid emulsion. Vitamins are added to the lipid
emulsion or to the bag immediately before use.
A number of other difficulties may be encountered. Polyunsaturated acids
are subject to hydrolysis. Any residual air might cause oxidation of labile
vitamins, e.g. vitamin C. Lipids (the fat emulsions and fat-soluble vitamins
formulated as an emulsion) may extract plasticizer from a plastic container,
especially if the bag is based on PVC. Any electrolytes may compromise emulsion
stability by altering the electro-chemistry around the dispersed oil droplets,
thus allowing the droplets to move closer to each other (due to a disruption of
the Stern layer) and coalesce; a less noticeable problem would be changes in
the globule size. Additionally, the plastic bag might absorb the oil-soluble
vitamins and care has to be taken in the selection of the container to avoid
moisture loss. As a final example of the complex nature of TPN fluids, amino
acids may undergo the Maillard reaction with glucose, resulting in
discoloration. An account of the clinical aspects of TPN can be found in Harper
& Lamerton (2009).
C) Small-Volume Injections
This category includes single-dose injections, usually of 1–2 ml but as
high as 50 ml, dispensed in borosilicate glass ampoules, plastic (polyethylene
or polypropylene) ampoules or, rarely, multiple-dose glass vials of 5–25 ml
capacity stoppered with a rubber closure through which a hypodermic needle can
be inserted, e.g. insulins, vaccines. The closure is designed to reseal after
withdrawal of the needle. It is unwise to include too many doses in a
multiple-dose container because of the risk of microbial contamination during
repeated use. Preservatives must be added to injections in multiple-dose
containers to prevent contamination during withdrawal of successive doses.
However, preservatives may not be used in injections in which the total volume
to be injected at one time exceeds 15 ml. This may occur if the solubility of a
drug is such that a therapeutic dose can only be achieved in this volume of
solvent. There is also an absolute prohibition on the inclusion of
preservatives in intra-arterial, intracardiac, intrathecal or subarachnoid,
intracisternal and peridural injections, and various ophthalmic injections.
i) Small-volume oily injections
Certain small-volume injections are available where the drug is
dissolved in a viscous oil because it is insoluble in water and therefore a
non-aqueous solvent is used. In addition, drugs in non-aqueous solvents provide
a depot effect, e.g. for hormones. The intramuscular route of injection must be
used. The vehicle may be a metabolizable fixed oil such as arachis oil or
sesame oil (but not a mineral oil) or an ester, such as ethyl oleate, which is
also capable of being metabolized. The latter is less viscous and therefore
easier to administer, but the depot effect is of shorter duration. The drug is
normally dissolved in the oil, filtered under pressure and distributed into
ampoules. After sealing, the ampoules are sterilized by dry heat. A
preservative is probably ineffective in such a medium and therefore offers very
little protection against contamination in a multiple-dose oily injection.
D) Freeze-Dried Products
In brief, freeze-drying (lyophilization) consists of preparing the drug
solution (with buffers and cryoprotectants), filtering through a bacteria-proof
filter, dispensing into containers, removing water in a freeze-drier, then
capping and closing the containers. Many biotechnology products are
freeze-dried.
Freeze-drying is an aseptic process whereby water is removed from a
frozen product mainly by sublimation, i.e. by the conversion of ice directly
into the vapour state without the intermediary of liquid water. It is a batch
process, of relatively long duration, and is used frequently for drugs of poor
stability. Drugs are reconstituted into solution immediately prior to injection.
The process consists of three stages:
·
freezing, which slows down degradation and solidifies the
product
·
primary drying, whereby energy is provided to the system and a vacuum applied to
expedite the removal of moisture at sub-ambient temperatures
·
secondary drying, whereby the product is heated to remove the last traces (2%) of water.
A number of characteristics of the
formulation control the behaviour of the product during the lyophilization
cycle. These include the glass transition temperature and the collapse
temperature. The maintenance of sterility and retention of the
appropriate sterility assurance level (SAL)
is implicit in the freeze-drier design (Pikal, 2007). Although
membrane-filtered sterile solutions may be used to fill containers to be placed
into the freeze-drier, other measures to maintain sterility are also employed.
These include using steam sterilization of the drier; gaseous sterilization has
not been widely adopted. The temperature of shelves is regulated using a
circulating fluid such as dimethyl-siloxane oil. Electronics and
computerization have led to the accumulation of better data for validation.
Stoppering systems allow the successful sealing of the containers and gas
entering the drier may be filtered to effect sterilization.
E) Packaging, Closures And Blow-Fill Technology
The packaging and closures must prevent loss of vehicle, excipient or
drug during sterilization and storage. Additionally, ingress of microorganisms
must be prevented. The packaging must not contribute any significant amounts of
extractable chemicals to the contents, e.g. vulcanizing agents from rubbers or
plasticizers from PVC infusion containers.
i) Glass
containers
Single-dose injections are usually packed in glass ampoules containing
1, 2 or 5 ml of product. To ensure removal of the correct dose volume by
syringe and needle, it is necessary to add an appropriate overage to the
ampoule. Thus a 1 ml ampoule will actually contain 1.1 ml of product and a 2 ml
ampoule should contain 2.15 ml of product.
Many injectables are sealed with a rubber closure held on by an
aluminium screw-cap or crimp-on ring. The rubber should be non-fragmenting, not
release soluble extractives, and be sufficiently soft and pliable to seal
around the needle inserted immediately prior to use. Although filled bottles
are sterilized by autoclaving, it is still possible for the infusions in glass
bottles to become contaminated with microorganisms through the seal before use.
For instance, during the final part of the autoclave cycle, bottles may be
spray-cooled with water to hasten the cooling process. However, if there is a
poor fit between bottle lip and rubber plug (a skirted inset type is used) it
is possible for the spray-cooling water to spread by capillary movement between
bottle thread and screw-cap and even to enter the bottle contents. Failure may
also result from any imperfection of the bottle or plug. Microorganisms may
gain access to the product within the containers during storage if hairline
cracks (due to bad handling or rough treatment) are present which permit fluid
seepage. Finally, contamination may occur during use (1) if poor aseptic
techniques are applied when setting up the infusion, (2) via an ineffective air
inlet (which allows replacement of the infused fluid with air in glass
bottles), or (3) when changing the giving set or bottle.
Three types of glass are suitable for use in the manufacture of
containers for injectable preparations. These are a neutral borosilicate glass,
a sulphated soda glass and a soft, moderately hydrolytic resistant glass. The
glasses are classified by their hydrolytic resistance. The choice for a
container depends on the properties of the solution they are used to package.
The advantages of glasses as container materials include their chemical resistance,
the fact that they do not absorb or leach organic materials, their impermeability
to water vapour and other gases, their transparency, their ability to form
rigid strong stable containers which resist puncture, their ability to hold a
vacuum and their overall stability to moist heat or dry heat sterilization.
However, glass containers may break and crack during the sterilization process,
they are attacked by alkaline solutions (and so may be a problem with, for
example, sodium citrate bladder irrigation), they are heavy and require venting
during administration of their contents.
ii) Closures
Closures are made of a polymer and their formulation include curing
agents, activators, antioxidants, plasticizers, fillers and pigments. They have
to be selected with the drug product in mind to avoid chemical incompatibility
and possible reaction with the ingredients in the product formulation. Sorption
of the preservative from multiple-dose formulations has frequently been a
problem and closures may therefore require saturation with the ingredients in
the product prior to packaging.
Closures should be flexible, to conform to the shape of the vial;
resilient, so as to reseal after each needle puncture; tough, so that low
fragmentation levels occur when punctured; non-thermoplastic, so that the heat
sterilization process is tolerated; and chemically compatible with the drug
formulation. Early closures were sulphur-based, and easily cured with
accelerators to speed up the curing rate. Unfortunately, a high degree of
water-extractable by-products could be taken up by the product that they were
intended to protect. Consequently they have been replaced by modern polymer
formulations with low extract curatives. Bromobutyl and chlorobutyl rubbers
show superior performance although special polymers, e.g. nitrile rubbers, are
used for mineral oil products. Problems of incompatibility may be overcome by
film bonding a fluorocarbon barrier film to the surfaces of the closures.
iii) Plastic containers
Most infusions are now packed in plastic containers. The plastic
material should be pliable, thermoresistant, transparent and non-toxic. The
plastics may contain antioxidants, stabilizers, lubricants, plasticizers,
fillers and colorants. Suitable materials are PVC (which may present a problem
with moisture loss) and polyethylene. The former is transparent and very
pliable, allowing the pack to collapse as the contents are withdrawn
(consequently no air inlet is required). These packs are also amenable to the
inclusion of ports into the bag, allowing greater safety during use. Such ports
may be protected by sterile overseals.
Two problems arise: (1) the possibility of toxic extractives, e.g.
diethyl phthalate, from the plastic entering the fluid if poor quality PVC is
used, and (2) moisture permeability leading to loss of water if the packs are
not protected by a water-impermeable outer wrap. Bags of high-quality
polyethylene are readily moulded (although separate ports cannot be included),
translucent and free from potential toxic extractives. Again, these packs
normally collapse readily during infusion. An important advantage of all
plastic packs is that the containers are hermetically sealed prior to
autoclaving and therefore spray-cooling water cannot enter the pack unless
there is seal failure, an easily detected occurrence. However, autoclaving of
plastic bags is more complex than that of bottled fluids because a steam/air
mixture is necessary to prevent bursting of the bags when heated (air
ballasting); adequate mixing of the steam and air is therefore required to
prevent layering of gases inside the chamber.
iv) Blow-fill technology
Blow-fill technology is an aseptic
process whereby the container is formed from thermoplastic granules, filled
with sterile solution and sealed, all within one automatic operation. The bulk
solution should have a low bioburden and is delivered to the machine through a
filling system that has been previously sanitized and steam sterilized in situ. Concern has been expressed that the machine
itself may generate particles. The plastic granules are composed usually of polyethylene,
polypropylene or one of their copolymers and are heat extruded at about 200°C
into a tube. The two halves of a mould close around this tube and seal the
base. The required quantity of sterile fluid is filled into the container,
which is then sealed. Products packed in this way include intravenous
solutions, and small-volume parenteral, ophthalmic and nebulizer solutions. The
technique offers lower costs than conventional packaging.
v) Cartridges and ready-to-use syringes
Small-volume injections may also be
packaged in cartridges or directly into disposable syringes. The latter are
immediately available for use but have a high cost of production and their
fixed content may lead to waste of material that remains un-injected after
single use. Cartridges are lower cost and may be fitted into injection pens;
many insulin products are produced in this manner because of their low waste,
ease of use and not requiring the patient to draw a dose volume into a separate
syringe. Cartridges have a plunger stopper at one end of a cylindrical glass
body containing the product for injection, and the other end is sealed with a
rubber-lined crimp cap. Processing steps include preparing the bulk sterile
solution for injection, washing and siliconizing the plunger stoppers, caps and
glass cartridges, inserting the plunger stopper, filling and closing. The
product is then sterilized, but care has to be taken that the internal
pressures that develop during the autoclave cycle do not force the cartridge
plunger out of the cartridge. The industry is developing a range of devices
designed to breach the skin’s defences to allow transdermal delivery
(Arora et al., 2008). These include microneedle arrays and
needle-free injections.
F) Quality Control Of Ampoules And Infusion Containers
i) Particulate contamination
Because of the possible clinical
consequences (such as granuloma of the lung) of injecting solid particles into
the bloodstream, the number of particles present in injections and other solutions
used in body cavities must be restricted. The British Pharmacopoeia (2010)
states that injectable preparations which are solutions ‘when examined under
suitable conditions of visibility are clear and practically free from particle’.
It also sets limits for sub-visible particles in injections based on the
principle of light blockage. Not more than 100 particles/ml greater than 5 μm
and not more than 50 particles/ml greater than 10 μm should be generally
obtained. The British Pharmacopoeia (2010)
describes a microscopic method for determination of the particulate
contamination of injections and intravenous infusions. The counting methods
should estimate extraneous particles, but not bubbles, that are unintentionally
present in the solutions. If the method provides a means for identifying and
detecting the particles, insight may be gained into their possible origin.
Filtration and observation using light microscopy have clear advantages,
including simplicity and allowing the operator to visualize the particles.
All parenterally injected solutions should be checked for particulate
contamination, but the above procedure is clearly impractical as a bulk
screening exercise. Those products contaminated with particulate matter should
be rejected. In practice, all products may be tested individually by a human
observer against split white/black screens and/or under polarized light for
obvious particulate contamination, and again there is a method described in
pharmacopoeias based on the split-screen technique. Nowadays optical control
equipment can take over this arduous and boring task.
ii) Integrity of
seals
The integrity of sealing of ampoules should be assessed on an individual
basis. Two techniques are available that depend on dye ingress under vacuum or
electronic means. With dye intrusion, the ampoules are submerged in a dye
solution and under an applied vacuum. Any container that has cracks in its
structure or is not sealed will admit the dye when the vacuum is reduced. On
washing, badly sealed ampoules will be coloured. This technique underestimates
the problem of bad sealing. In the alternative technique, high-frequency spark
testing, the presence of a leak causes a change in a high-frequency electrical
signal placed across the ampoule. The method is limited to aqueous products
with a high conductivity. It is a very sensitive technique and detects weak
seals not detected by the dye test. In reality, both tests should be used in
parallel.
Related Topics
