Manufacture of Drugs
| Home | | Forensic Pharmacy |Chapter: Forensic Pharmacy : The Drugs and Cosmetics Act (DCA) 1940 and Rules 1945
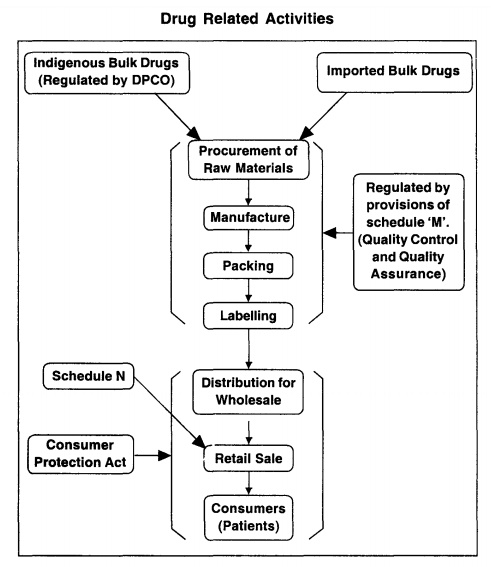
Manufacture in relation to any drug includes any process or part of a process for making, altering, finishing, packing, labelling, breaking up or otherwise treating or adopting any drug with a view to its sale and distribution, but does not include compounding or dispensing of any drug or packing of any drug in ordinary course of retail business.
Manufacture of Drugs
Definition: Manufacture in
relation to any drug includes any process or part of a process for making,
altering, finishing, packing, labelling, breaking up or otherwise treating or
adopting any drug with a view to its sale and distribution, but does not
include compounding or dispensing of any drug or packing of any drug in
ordinary course of retail business.
Manufacture
of drugs is a blend of art and science, to be achieved strictly in accordance
with the provisions of Good Manufacturing Practices (GMP). A person who is
interested in starting manufacturing of drugs is required to fulfill several
conditions laid down in DCA and Rules. The conditions to be fulfilled before
licence is granted are collectively called as "Conditions Precedent"
and conditions that are required to be fulfilled after the licence is obtained
for manufacturing are called "Conditions Subsequent". The Licensing
Authority is both in States and at Central Government. The Central Government
is empowered to prohibit manufacturing and sale of any drug formulation in
public interest.
Licences are
required for the manufacturing of following categories of drugs.
1. Manufacturing of drugs belonging to Schedules C and C (I)
2. Manufacturing of drugs belonging to Schedule X
3. Manufacturing of drugs belonging to Schedules C, C (I)
and X
4. Manufacturing of drugs other than Schedule C, C, and X
5. Manufacturing of drugs for examination, test or analysis
6. Loan licences
7. Licence for Repacking
The following
categories of drugs and cosmetics are prohibited to be manufactured or sold in
our country.
1. Any drug or
cosmetic which is substandard, misbranded, adulterated or spurious.
2. Any patent or
proprietory medicine without clear indication of ingredients.
3.
Any drug claiming for accurate cure or prevention of diseases listed in
Schedule].
4. Any manufacturing
of formulation containing drug or cosmetic which has been imported into our
country in contravention to the provisions of the Act and Rules.
5. Manufacturing for
sale of any drug or cosmetic containing any harmful ingredient.
6. Manufacturing for sale of any drug or cosmetic in
contravention to the provisions of the Act and Rules, provided that manufacture
of small quantities of any drug for the purpose of examination, test or
analysis is permitted, subject to prescribed conditions.
Separate
applications for separate licences for more than one premises of manufacture
are required to be made.
(A) Manufacturing of Drugs other than Schedules C and C (1)
Application for
grant of licence or renewal is made in Form 24. The licence is issued by
Licensing Authority in Form 25.
I - Conditions for grant of Licence are as follows:
1. Competent Staff: (i) A graduate in
Pharmacy/Pharmaceutical Chemistry
with a minimum of 18 months of
experience after graduation. The duration may be reduced by 6 months, if the
applicant has undergone training during graduation. or (ii)A graduate in
Science with Chemistry as a principal subject and with 3 years of manufacturing
experience after graduation or (iii) A graduate in Chemical Engineering or
Chemical Technology or Medicine with 3 years of manufacturing experience after
graduation or (iv) holding equivalent foreign qualification.
For disinfectant
fluids. insecticides, liquid paraffin, non-chemical contraceptives, surgical
dressings, medicinal gases, plaster of paris, only adequate experience in
manufacturing is required and no specific qualification is mentioned
2. Factory premises: As per Schedule' M' with regard to
premises, space, plant and
equipment.
3. Separate facilities for analysis of raw materials
and finished formulations
There should be
separate head of department for analysis and manufacturing sections. Head of
the testing and analytical department should be a graduate in
Medicine/Pharmacy/Pharmaceutical Chemistry/Science with adequate experience in
analysis.
4. There should be
adequate arrangement of storage of raw materials and finished products.
5. While applying for
licence to manufacture patent or proprietary medicines, it is required to
submit evidence justifying therapeutic claims of the product, its stability and
safety.
II. Conditions to be fulfilled after getting a licence:
1. The manufacturer
should always maintain adequate staff, sufficient premises and equipment.
2. The manufacturing
records, records for raw material and analysis and other operational records
should be maintained as per Schedule 'U'.
3.
The licensee should own an analytical laboratory or get tested the samples
analysed in an approved analytical laboratory.
The records for
analysis are required to be maintained for a period of 5 years from date of
manufacturing. The records are both for manufacturing and for finished product.
4. The manufacturer
should allow the Inspector to inspect the premises, manufacturing process,
analytical procedures, and withdraw the samples. The samples may be provided on
demand and entire protocol of manufacturing should be made available when asked
for. The manufacturer should also withdraw the batch manufactured by him if
directed to do so by the Controlling Authority.
5. The manufacturer
should comply with all the requirements of the Act and Rules thereunder.
6. The manufacturer should maintain the Inspection Book.
7. Samples with expiry date should be maintained for 6
months after expiry date. For other categories, the samples should be
maintained for 3 years from the date of manufacturing. Twice the lot of
reference samples should be maintained. The quantity maintained should be
sufficiently available for analysis.
8. Any change in the
staff structure especially, technical staff should be reported to Licencing
Authority.
9. Any major
structural change in the premises should be done with the permission of
Licencing Authority
10. The manufacturer
should forward all the sales records to the Controlling Authority.
[B] Manufacture of Drugs for Testing, Analysis or Examination
If
the manufacturer does not hold separate licence for test, analysis or
examination, the licence is obtained in Form 29. The provisions relating prohibition
of manufacturing of certain drugs do not apply for such manufacturing meant for
test or analysis. The validity of the licence is for I year. The manufactured
drugs should be kept in containers bearing appropriate label indicating the
purpose of test or analysis. When the material is supplied to other
manufacturer, the label stating the name and address of manufacturer,
scientific name of the drug, licence number, date of manufacture, etc., should
be provided.
The
manufacturer should allow the Inspector to inspect the premises, manufacturing,
and analytical records and withdraw the samples if required for analysis. The
manufacturer should comply with the provisions of the Act and Rules. The manufacturer
should maintain an Inspection Book and the same be shown to the Inspector.
[C] Manufacturing of New Drugs
In addition to
provisions for manufacture of drugs, there should be documentary evidence for
qual ity, purity, therapeutic trials of new drugs and evidence for approval
under schedule 'Y' (Clinical trials).
[D] Loan Licences
For
drugs other than Schedules C, (I) and X, loan licences can be given. A
qualified person can make use of approved facilities of manufacturing provided
by any other person and obtain a loan licence for manufacture of drugs other
than Schedules C, C (I) and X. The licensee in such cases should convice the
Licensing Authority about the availability of the infrastructure on loan from
approved manufacturer, and also be convinced about the need to grant such a
loan licence.
All
the conditions of GMP in Schedule 'M' are required to be fulfilled.
Manufacturing Records should be maintained for 5 years. In case of drugs with
date of expiry, the records should be maintained for 2 years. Application for
grant or renewal of loan licence is made in Form 24-A. The licence is issued by
Licensing Authority in Form 25-A, which is valid for I year.
[E] Repacking licence
It is issued for drugs
other than Schedules C, C1 and X, subject to fulfillment of conditions. The
application is made for grant or renewal of licence in Form 24-B. The licence
is issued by Licensing Authority after inspection in Form 25-B.
There should be
adequate arrangement for testing of samples. The licence should always be
displaced at premises of repacking. The factory primises for repacking should
comply with provisions of Schedule M. Hygienic conditions of working should
always be maintained. Adequate staff should be appointed and any change in
staff structure should be immediately informed to Controlling Authority.
The
Competent person eligible to get Repacking Licence is -
1. Diploma in pharmacy or Registered pharmacist
2. Intermediate with Chemistry as a subject
3. Matriculation
with 4 years of experience in manufacture, dispensing or repacking of drugs.
The licence is valid
till 3st December every year and required to be renewed. There should be
separate application for separate licence.
The
container or package of repacked drug should bear on its label the words - "Rpg.Lic.No".
The Repacking Licence is given to the competent persons for the bulk drugs or
drug formulations procured in large quantities from manufacturer directly and
required to be sold in small quantities.
[F] Manufacture of Drugs under Scheaules C and C (1) and Drugs specified in Schedules C, C (1) and X
Application
for grant or renewal of a licence is made for Schedules C and C (1) drugs in
Form-27 and for Schedules C, C (1) and X in
Form 27-B. The licence is
issued in Form-28 for manufacture of
Schedules C and C (1) drugs and in Form
28-B for drugs under Schedules C, C (I) and X.
1. The conditions
stipulated in Schedule 'M' should be met with. The requirements of space,
plant, equipment, etc., should be as per Schedule M.
2. The testing
strength or quality strength of the manufacturing un it should be assessed.
There should be a qualified independent head for analytical wing including for
analysis of Schedules C and C (1)
drugs.
3. The competent technical persons should be available with
one of the following qualifications.
(i) A graduate in
Pharmacy/Pharmaceutical Chemistry with 18 months of experience after graduation
in manufacturing drugs to which licence applies. Six months of mandatory
training after graduation can form part of this total training of 18 months.
or
A graduate in
Science with Chemistry or Microbiology as major subject with a minimum of 3
years of experience after graduation in the manufacture of drugs to which this
licence applies.
or
A foreign equivalent
qualification or an expert in manufacturing before 29th June, 1957.
For
manufacture of veterinary Scheduled drug formulations, a graduate in
Medicine/Veterinary Science/General Science/ Pharmacy with minimum 3 years
experience in manufacture of biological products.
4. There should be
facilities available for storage of drugs. Sufficient precaution should be
taken for preserving the properties of manufactured drugs.
5. Records of
manufacturing should be maintained as per Schedule 'U'. Records of test, analysis
and examination and batch wise records are required to be maintained. All
records should be kept for a period of 2 years from the date of expiry of drug.
In other cases, with no expiry date, the records should be kept for 5 years
from the date of manufacturing.
6. The manufacturer
should allow the Inspector or the representative of Controlling Authority and
Licensing authority to visit manufacturing premises with or without notice.
7. The manufacturer
should report to the Licensing Authority any changes in staff or any material
changes.
8. The licensee should furnish the sample of drugs to the
Inspector when asked for purpose of analysis. He should not sell the drug if
asked to do so by Controlling Authority and withdraw the drug already sold from
the market on direction from the Authority
9. The licensee
should maintain the Inspection Book to enable the Inspector to record his
observations.
Related Topics
