Patterns of Cutaneous ADRS
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Dermatological ADRs
Exanthematous or maculo-papular eruptions, often reported as ‘drug rashes’ or ‘drug eruptions’, are the most common ADRs affecting the skin.
PATTERNS OF CUTANEOUS ADRS
EXANTHEMATOUS DRUG ERUPTION
Exanthematous
or maculo-papular eruptions, often reported as ‘drug rashes’ or ‘drug
eruptions’, are the most common ADRs affecting the skin. The main mechanism is
probably immunologic, and may corre-spond to type IV delayed cell-mediated
hypersensi-tivity reaction.
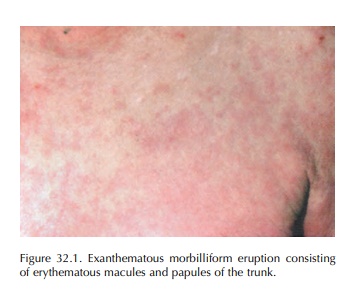
The eruption usually occurs between 4 and 14 days after beginning a new therapy, and even a few days after it has ceased (‘eruption of the ninth day’). However, it can develop sooner, especially in the case of rechallenge. The eruption consists of erythematous macules, papules, often symmetric. They begin on the trunk, upper extremities, and progressively become confluent (Figure 32.1). The eruption is typically poly-morphous: morbilliform or sometimes urticarial on the limbs, confluent on the thorax, purpuric on the feet. Mucous membranes are usually not involved. Pruri-tus and low-grade fever are often associated with the eruption, which frequently lasts less than 2 weeks.
Cutaneous
pathological slides exhibit a very mild lymphocytic infiltrate around vessels
of the dermis, and a few necrotic keratinocytes within the epider-mis. This
pattern, often difficult to differentiate from normal skin is not specific, and
cannot help to distinguish a drug eruption from an eruption of another cause.
The
differential diagnosis of exanthematous drug reactions includes viral eruptions
(EBV, CMV, HHV6, Parvovirus B19, etc.), toxinic eruptions, acute Graft-vs-Host
reaction, Kawasaki syndrome, Still’s disease, and so on. Dermatologists usually
consider that viral infections are the cause of most drug erup-tions in
children, while drugs are more frequently responsible in adults.
Treatment
is largely supportive, usually after the removal of the offending agent,
associated with topi-cal corticosteroid and systemic antipruritic agents. When
the suspected drug is of paramount importance for the patient (e.g.
antibacterial sulphonamides in AIDS patients) treating ‘through the eruption’
can be considered as an option. In most instances, the erup-tion will disappear
in about the same time as if the drug had been withdrawn. Because a few
patients may experience a progressive worsening of the eruption leading to one
of the severe reactions described below, the benefit–risk ratio of this
attitude should be care-fully weighted and the evolution of the rash strictly
monitored.
Most
drugs can induce an erythematous eruption in about 1% of users. The following
drugs have higher risks (more than 3% of users): allopurinol,
aminopeni-cillins, cephalosporins, antibacterial sulphonamides and most
antiepileptic agents.
URTICARIA AND ANGIO-OEDEMA
Urticaria
is a common, transient eruption of erythe-matous and oedematous papules and
plaques, usually associated with pruritus. When dermal and subcuta-neous
tissues are involved, this reaction is known as angio-oedema. Most cases of
angio-oedema are associated to urticaria. They can be complicated by a life-threatening
anaphylactic reaction. Urticaria, angio-oedema and anaphylaxis may be a type I
hypersensitivity reaction mediated by IgE antibodies (penicillin allergy). But
other ‘anaphylactoid’ mecha-nisms, leading to direct and non-specific
liberation of histamine or other mediators of inflammation, are also common for
drug reactions (contrast media, NSAIDs including aspirin).
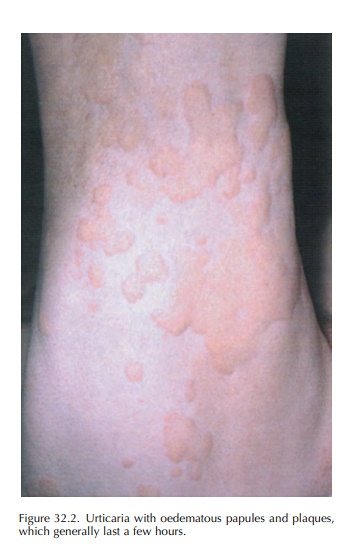
Clinically, itchy erythematous, oedematous papules and plaques develop in variable numbers and size (Figure 32.2). They are localized anywhere on the body, including the palm, soles and scalp. They frequently last a few hours and disappear within 24 hours, leaving the skin with a normal appear-ance. Angio-oedema is often associated with urticaria, consisting of pale or pink swellings which affect the face (eyelids, lips, ears, etc.) but also buccal mucosa, the tongue, larynx, pharynx, and so on. More severe reaction, such as anaphylaxis, can involve other systems and lead to respiratory collapse, shock and eventually death.
Urticaria is histologically non-specific with a super-ficial
and deep scarce infiltrate of mononuclear cells accompanied by eosinophils and
neutrophils, oedema-tous reticular dermis, vascular and lymphatic dilata-tion.
The epidermis is uninvolved.
Urticaria has been classified into acute, when the eruption
lasts less than 6 weeks, or chronic when it persists much longer.
It usually occurs within a few hours of drug
admin-istration, but may also occur within a few minutes.
Withdrawal
of the causative agent is the main treat-ment. It can sometimes be associated
with histamine H1 receptor blockers. Systemic steroids and an
intra-muscular injection of epinephrine are necessary in an emergency if severe
angio-oedema and anaphy-laxis occur.
Many
drugs can induce urticaria (most often of the acute type), but more than 80% of
cases of urticaria are related to other causes (stings, food allergy, etc.).
Antibiotics, especially penicillin, and general anaes-thetics are classic causes
of IgE-mediated hypersen-sitivity reaction. A radioallergosorbent test (RAST)
or ELISA and skin tests (prick-tests) can be useful to confirm the diagnosis.
Because they may rarely induce an anaphylactic reaction, prick-tests must be
performed only by experienced physicians.
The
two most frequent causes of drug-induced non-IgE-mediated urticaria and
angio-oedema are NSAIDs and angiotensin-converting enzyme (ACE) inhibitors.
Angio-oedema occurs in 2 to 10 per 10 000 new users of ACE inhibitors (Hedner et al., 1992), a rate that is probably
higher than the risk associated with penicillins (about 1 per 10 000 courses).
The reac-tion begins much later than IgE-mediated urticaria, usually in the
first weeks of treatment. Up to one-third of patients with angio-oedema related
to ACE inhibitors have a recurrence when using angiotensin 2 receptor
antagonists (van Rijnsoever et al.,
1998). This suggests a pharmacologic mechanism.
PHOTOSENSITIVITY
Cutaneous
photosensitivity diseases may be idio-pathic, produced by endogenous
photosensitizers (e.g. porphyrins) or associated with exogenous
photo-sensitizers like drugs. The association of light and a drug can be
responsible for acute inflammation of the skin. The photosensitivity reactions
are divided into two types: phototoxicity and photoallergy (Gould, Mercurio and
Elmets, 1995).
PHOTOTOXICITY
Phototoxic
disorders are not rare and always predictable. It can occur in any person who
receives sufficient quantities of a phototoxic drug, together with the proper
light exposure. The reaction results directly from photochemistry involving the
skin. The association of light with a photosensitizing chemical in the skin
creates an unstable singlet or triplet state within the electrons. This leads
to the generation of reactive oxygen, which is responsible for cell damage.
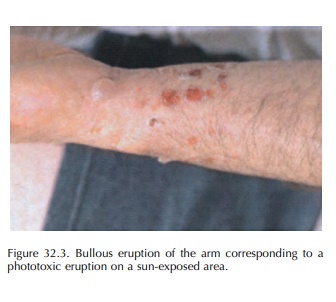
Clinical
manifestations usually present as an exag-gerated sunburn occurring in
sun-exposed areas only (Figure 32.3). This is followed by hyperpigmentation.
Photo-onycholysis and pseudoporphyria (blisters on sun-exposed parts of the
limbs) are less common clin-ical forms.
Phototoxicity
is histologically characterized by epidermal cell degeneration with necrotic
keratinocytes, oedema, sparse dermal lymphocytic infiltrate and vasodilatation.
Phototoxicity is easily documented in
vitro or in vivo. A photopatch
test will be positive in all individuals and will therefore not be a
discriminator for causality assessment. The minimal dose of UV (UVA more often
than UVB) inducing an erythema will be decreased in all subjects during
treatment.
PHOTOALLERGY
A
photoallergic reaction is considered as a result of cell-mediated
hypersensitivity. Ultraviolet radiation is required to convert a drug into an
immunopatholog-ically active compound (photo-antigen) that induces the immune
response.
Photoallergic
eruption is more chronic than photo-toxicity and is mainly eczematous and
pruritic. A lichen planus-like reaction has also been reported. It is usually
more marked in exposed sites, but may often progress outside these areas. In the
chronic phase, erythema, scaling and lichenification predom-inate.
Photoallergic reactions are usually transient and resolve after a variable
length of time when the offending agent has been removed. Rarely, an extreme
sensitivity to sun may persist for months or years (‘persistent light
reactors’). Photopatch testing is valuable when photoallergy is suspected. A
multitude of drugs induce photoallergic reactions, includ-ing antibiotics
(sulphonamides, pyrimethamine, fluo-roquinolones), fragrances, NSAIDs,
phenothiazine, thiazide diuretics, and so on.
In
phototoxic reactions, the treatment requires removal of the offending agent
and/or avoidance of sun exposure. For a drug with a short elimi-nation
half-life, administration in the evening may be enough to decrease the risk
below the clinical threshold. In photoallergy, drug withdrawal is recom-mended,
because of the risk of worse reactions even with low UV doses. Topical
corticosteroid, systemic antipruritic agents may be useful.
VASCULITIS
Vasculitis
corresponds to immune-mediated inflam-mation and damage to a blood vessel’s
wall. It may be caused by a variety of agents, especially infec-tions and
collagen vascular diseases. Many cases remain idiopathic. Drug-induced
vasculitis is believed to result from antibodies directed against drug-related
haptens (Roujeau and Stern, 1994). Direct drug toxi-city against a vessel’s
wall, autoantibodies reacting with endothelial cells and cell-mediated
cytotoxic reactions against vessels were also proposed as expla-nations. The
precise mechanism is still unknown.
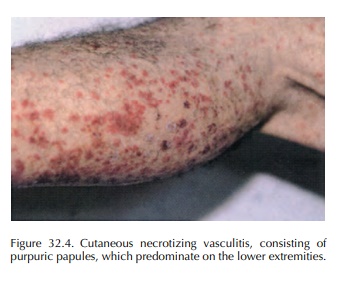
This drug-induced eruption corresponds to a cuta-neous necrotizing vasculitis consisting of palpa-ble purpuric papules which predominate on the lower extremities (Figure 32.4). Urticaria-like lesions, ulcers, nodules, hemorrhagic blisters, Raynaud’s disease and digital necrosis may also occur. The vasculitis may involve other organs, with fever, arthralgias, myalgias, headache, dyspnea, neurologi-cal involvement and renal abnormalities, sometimes life-threatening. The histology of small blood vessels exhibits necrotizing and/or leukocytoclasic vasculitis. The direct immunofluorescence is often positive, with immunoglobulin and C3 deposits on capillary walls.
Vasculitis
occurs 7 to 21 days after drug adminis-tration, and less than 3 days after
rechallenge. With-drawing the drug usually leads to a rapid resolution. A
systemic corticosteroid may benefit some patients.
Drug-induced
cases are a minority of cases of vasculitis (no more than 10% in a large
series) and have to be differentiated from other causes of cutaneous
vasculitis: infection, autoimmune diseases (polyarteritis nodosa, Wegener’s
granulomatosis, etc.), Schönlein-Henoch purpura and cancer.
The
main drugs implicated are allopuri-nol, NSAIDs, cimetidine, penicillin,
hydantoin, sulphonamides and propylthiouracil.
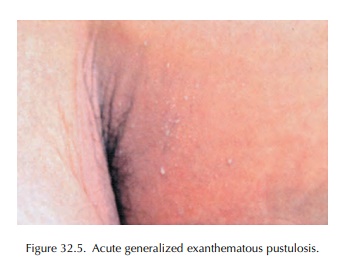
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
In 1980, Beylot et al. described an acute pustular dermatosis named ‘Acute generalized exanthematous pustulosis’ (AGEP) (Beylot, Bioulac and Doutre, 1980). Of these eruptions at least 80% could be drug-induced. Hypersensitivity to mercury and infection with enteroviruses may also be responsible. The inci-dence of AGEP has been under-estimated and many cases have been confused with pustular psoriasis. Synonyms are pustular drug rash, pustular eruption and pustuloderma (Staughton et al., 1984). Proposed diagnosis criteria (Roujeau et al., 1991) include:
• an acute pustular eruption;
• fever above 38 C;
• neutrophilia with or without a mild eosinophilia;
• subcorneal or intraepidermal pustules on skin
biopsy;
• spontaneous resolution in less than 15 days.
AGEP
is characterized by fever, which generally begins the same day as the pustular
rash. Numer-ous, small, mostly non-follicular pustules arise on a widespread
oedematous erythema, burning pruritic or both (Figure 32.5). Oedema of the face
and the hands, purpura, vesicles, blisters, erythema multiforme-like lesions
and mild involvement of mucous membrane have also been associated. Pustules are
mainly local-ized on the main folds (neck, axillae, groins, etc.), trunk and
upper extremities.
The
histopathology shows spongiform pustules located under the stratum corneum, the
most superfi-cial layer of the epidermis. Papillary dermal oedema and
perivascular polymorphous infiltrate are usually present. Leukocytoclasic
vasculitis and focal necrotic keranocytes have also been reported.
Hyperleukocytosis
with elevated neutrophils count, transient renal failure and hypocalcemia are
frequently seen.
There
are two different times between the drug administration and the skin eruption.
For antibiotics it is usually very short, less than 2 days. A more clas-sical
delay of 1–2 weeks is observed with diltiazem, another classical inducer. The
eruption lasts 1 to 2 weeks, and is followed by a superficial desquamation. The
withdrawal of the responsible drug is the main treatment, associated with a
topical corticosteroid and sometimes a systemic antipruritic agent.
AGEP
must be differentiated from acute pustular psoriasis of the von Zumbusch type.
The pustules in both diseases are clinically indistinguishable; the
histopathology can be helpful.
Antibiotics
(béta-lactam, some macrolides and quinolones) are the main drugs implicated in
AGEP.
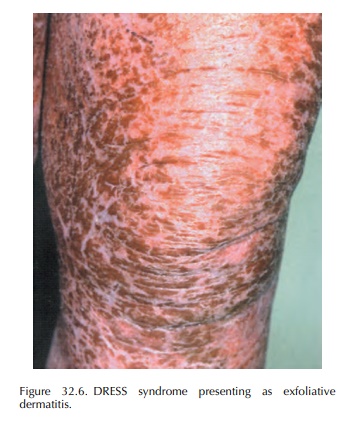
DRESS/HYPERSENSITIVITY
‘Hypersensitivity
syndrome’ refers to a specific severe skin reaction. The acronym of DRESS for
Drug Reaction with Eosinophilia and Systemic Symptoms has been proposed as more
specific than ‘hyper-sensitivity’, which would be appropriate for most types of
drug reaction. It has been estimated to occur in between one in 1000 and one in
10 000 exposures with drugs such as antiepileptics and sulphonamides. This
syndrome is typically charac-terized in its complete form by a severe eruption,
lymphadenopathy, fever, hepatitis, interstitial nephri-tis, pulmonary
infiltrates and sometimes arthralgias. The clinical lesions are associated with
haemato-logical alterations: eosinophilia and lymphocytosis with basophil
lymphocytes (Shear and Spielberg, 1988; Roujeau and Stern, 1994; Callot et al., 1996). Multivisceral involvement
differentiates hypersensi-tivity syndrome from common exanthematous erup-tion.
Some consider that Stevens–Johnson Syndrome (SJS) and toxic epidermal
necrolysis (TEN) may occur as part of a ‘hypersensitivity syndrome’. The skin
lesions and visceral complications are actually differ-ent. Eosinophilia and
atypical lymphocytosis are not observed in SJS and TEN.
These
reactions are more frequent among persons of African ancestry. They begin 2 to
6 weeks after the first drug use, later than most other skin reactions. Fever
and skin rash are the most common symptoms. Cutaneous manifestations begin as a
morbilliform rash, which later becomes infiltrated with an oede-matous
follicular accentuation (Figure 32.6). Erythro-derma, vesicles, tight blisters
induced by dermal oedema, follicular as well as non-follicular pustules can
also occur. Face, upper trunk and extremities are initially involved. Oedema of
the face is frequent and evocative of diagnosis.
Prominent
eosinophilia (70% of cases) and atypical lymphocytosis (50–60%) are the most
characteristic biological features of this reaction. Liver abnor-malities with
raised aminotransferase, alkaline phoshatase, bilirubin levels and abnormal
prothrombin time are present in about 50% of patients.
Histopathology
exhibits a rather dense lymphocytic infiltrate in the superficial dermis and/or
perivascular, associated with dermal oedema.
Rash
and hepatitis may persist for several weeks after drug withdrawal, and some of
the manifestations may be life-threatening.
The
differential diagnosis includes other cuta-neous drug reactions, acute viral
infection, idiopathic hypereosinophilic syndrome, lymphoma and pseu-dolymphoma.
Special attention should be paid to viral infection and specially to HHV6,
since several publi-cations suggest a possible interaction between DRESS and
reactivation of HHV6 or other lymphotropic viruses (Descamps et al., 2001; Kano, Inaoka and Shiohara,
2004).
Topical
high-potency corticosteroids can be help-ful in skin manifestations. Systemic
corticosteroids are often proposed when internal organ involve-ment exists.
The
aromatic antiepileptic agents (phenobarbi-tal, carbamazepine, phenytoin),
minocycline and sulphonamides are the most frequent causes of hyper-sensitivity
syndrome; allopurinol, gold salts and dapsone may also induce this syndrome.
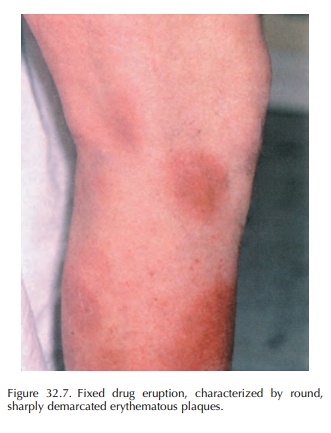
FIXED DRUG ERUPTION
A
fixed drug eruption is an exclusively drug-induced cutaneous reaction. The
lesions develop usually less than 2 days after the drug intake. Clinically,
they are characterized by a solitary or few, round, sharply demarcated
erythematous and oedematous plaques, sometimes with a central blister (Figure
32.7). The eruption can be located on every site of the body and may involve
mucous membranes, principally the lips and genitalia. The eruption
progressively fades in a few days, to leave a post-inflammatory brown
pigmentation. With rechallenge with the causative drug, the lesions recur at
exactly the same sites. After several relapses the eruption may involve large
areas of the body. This Generalized Fixed Drug Eruption may be difficult to
distinguish from TEN.
Histopathology
reveals a superficial and deep dermal and perivascular infiltrate (composed of
lymphocytes, eosinophils, and sometimes neutrophils) associated with necrotic
keratinocytes. Dermal macrophages pigmented by melanin (melanophages) when
present are considered an important clue to the diagnosis.
The
drugs most frequently associated with fixed drug eruption are phenazone
derivates, barbiturates, tetracycline, sulphonamides and carbamazepine
(Kauppinen and Stubb, 1984).
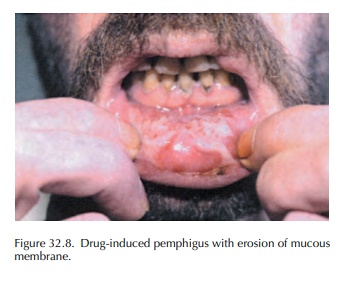
DRUG-INDUCED PEMPHIGUS
Pemphigus is a chronic autoimmune blistering disease provoked by autoantibodies reacting with normal constituants of desmosomes, the structures that provide attachment between epidermal cells. It presents clinically with flaccid intraepidermal blis-ters and erosions of the skin and mucous membranes (Figure 32.8). Nikolsky’s sign is found.
The
histology exhibits detachment of epidermal cells (acantholysis), responsible
for intraepidermal blisters located subcorneally (pemphigus foliaceus) or in
the lower epidermis (pemphigus vulgaris).
Direct
immunofluorescence performed to a perile-sional skin biopsy specimen reveals
immunoglobulin deposits around keratinocytes in the epidermis in all ‘spontaneous’
cases but in only 50% of drug-induced cases. The presence in the serum of
autoantibodies reacting against the epidermis is detected by indirect
immunofluorescence, Western-blot or ELISA tests.
In
Western countries up to 10% of cases of pemphi-gus could be drug-induced. It
begins several weeks or months after drug therapy is initiated. It presents as
pemphigus foliaceus or as pemphigus vulgaris with mucosal involvement. The main
drugs incriminated are d-penicillamine and other drugs containing a thiol
radical, like captopril and piroxicam. The remission after drug withdrawal is
not always spontaneous, particularly in cases of pemphigus attributed to drugs
that do not have a thiol part.
SJS AND TEN
SJS
and TEN are rare, life-threatening, drug-induced skin reactions. The incidence
of TEN is evaluated to 0.4 to 1.2 cases per million person-years and of SJS
from 1 to 6 cases per million person-years (Roujeau and Stern, 1994). The
immunopathologic pattern of early lesions suggests a cell-mediated cyto-toxic
reaction against epidermal cells. Widespread
apoptosis of epidermal cells is provoked by the activa-tion of several
pathways: the interaction of Fas antigen (cell surface death receptor) and Fas
ligand but also perforin plus granzyme and TNFalpha.
With
others we proposed to consider SJS and TEN as severity variants of the same
drug-induced disease, and to distinguish SJS from erythema multiforme major
(Bastuji-Garin et al., 1993), the
latter being mostly related to infections, especially with herpes (Auquier-Dunant
et al., 2002).
According
to this proposal, erythema multiforme (major when mucous membranes are
involved) is characterized by typical concentric ‘target’ lesions acrally
distributed, with limited blisters (detachment rarely involves more than 2–3%
of the body surface area). The pathology shows an interface dermatitis with
moderate to marked lymphocyte infiltrate in the dermis, exocytosis and mild
necrosis of epidermal cells. In our experience, erythema multiforme is rarely
drug-induced. Most of the cases that are reported or published as drug-induced
erythema multiforme are either cases that we would label as SJS or cases of
erythematous drug eruptions, because of confusion between ‘multiforme’ and the
polymorphous patterns of many erythematous eruptions.
SJS
is characterized by atypical targets and more often by small blisters arising
on purple macules. Lesions are widespread and usually predominate on the trunk.
Confluence of blisters on limited areas leads to detachment below 10% of the
body surface area. The pathology can be separated from that of erythema
multiforme by less lymphocyte infiltrate and more epidermal necrosis
(Wolkenstein et al., 1998).
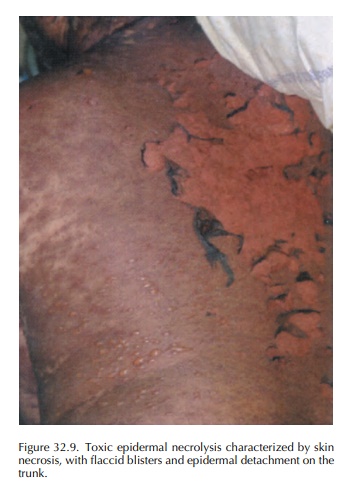
Toxic
epidermal necrolysis is characterized by the same lesions as SJS but with a
confluence of blis-ters leading to a positive Nikolski sign and to the
detachment of large epidermal sheets on more than 30% of the body surface area
(cases with detachment of between 10 and 30% are labelled overlap SJS-TEN)
(Figure 32.9). Skin pathology shows necrosis of full-thickness epidermis and
negative immunoflu-orescence. This is important for distinguishing TEN from
exfoliative dermatitis, staphylococcal scalded skin syndrome, acute
exanthematous pustulosis and paraneoplastic pemphigus, which may be misdiag-nosed
as SJS or TEN.
Patients
with SJS or TEN have high fever. Severe erosions of mucous membranes are nearly
constant.
Systemic
manifestations include mild elevation of hepatic enzymes (overt hepatitis in
10% of cases), intestinal and pulmonary manifestations (with slough-ing of
epithelia similar to what happens to the skin). Leucopenia is frequent and
eosinophilia unusual. Death occurs in 10% of patients with SJS and more than
30% of patients with TEN, principally from sepsis or pulmonary involvement (Roujeau
and Stern, 1994).
The
treatment is mainly symptomatic, consisting of nursing care, maintenance of
fluid and electrolyte balance and nutritional support. Early withdrawal of all
potentially responsible drugs is essential. Short courses of corticosteroids
early in the disease have been advocated, but their effectiveness has never
been demonstrated in controlled trials. Thalidomide has been shown to be
detrimental in TEN, possibly because of a paradoxical enhancement of TNFalpha
production. High-dose intravenous immunoglobulins were disappointing in our
experience.
Drug
reactions are responsible for at least 70% of cases of both SJS and TEN
(Knowles, Uetrecht and Shear, 2000). Antibacterial sulphonamides,
anticon-vulsants, oxicam and pyrazolone NSAIDs, allopurinol and chlormezanone
are the drugs associated with the higher risks. An international case–control
study of SJS and TEN found relative risks of between 50 and 172 for new users
(treatment duration of less than 2 months) of the above-mentioned drugs and
also for corticosteroids (Roujeau et al.,
1995). In that study, excess risks for associated drugs were in the range of 1
to 4.5 cases for 1 million users per week (Roujeau et al., 1995).
SJS
and TEN typically begin within 4 weeks of initiating therapy, usually 7 to 21
days after the first drug exposure and sometimes a few days after the drug has
been withdrawn. It occurs more rapidly with rechallenge.
Related Topics
