Pharmacokinetics
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacokinetics; Membrane Transport, Absorption And Distribution Of Drugs
Pharmacokinetics is the quantitative study of drug movement in, through and out of the body.The intensity of response is related to concentration of the drug at the site of action, which in turn is dependent on its pharmacokinetic properties. Pharmacokinetic considerations, therefore, determine the route(s) of administration, dose, latency of onset, time of peak action, duration of action and frequency of administration of a drug .
PHARMACOKINETICS
Pharmacokinetics is
the quantitative study of drug movement in, through and out of the body. The
overall scheme of pharmacokinetic processes is depicted in Fig. 2.1. The
intensity of response is related to concentration of the drug at the site of
action, which in turn is dependent on its pharmacokinetic properties.
Pharmacokinetic considerations, therefore, determine the route(s) of administration,
dose, latency of onset, time of peak action, duration of action and frequency
of administration of a drug .
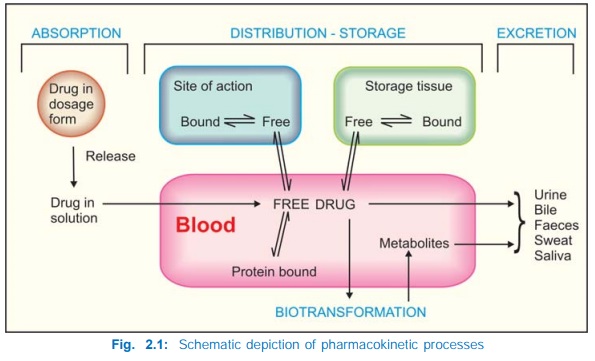
All pharmacokinetic
processes involve transport of the drug across biological membranes.
Biological membrane
This is a bilayer (about 100 Å thick) of phospholipid and cholesterol
molecules, the polar groups (glyceryl phosphate attached to
ethanolamine/choline or hydroxyl group of cholesterol) of these are oriented at
the two surfaces and the nonpolar hydrocarbon chains are embedded in the matrix
to form a continuous sheet. Extrinsic and intrinsic protein molecules are
adsorbed on the lipid bilayer.
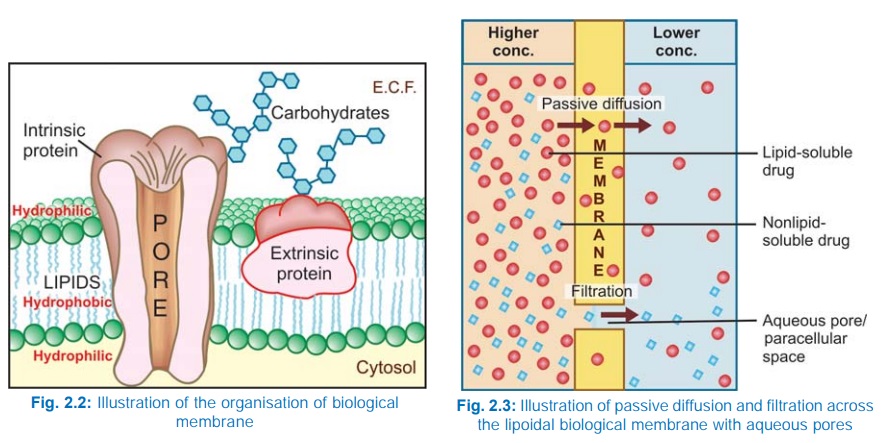
Glycoproteins or
glycolipids are formed on the surface by attachment to polymeric sugars,
aminosugars or sialic acids. The specific lipid and protein composition of
different membranes differs according to the cell or the organelle type. The
proteins are able to freely float through the membrane: associate and organize
or vice versa. Some of the intrinsic ones, which extend through the full
thickness of the membrane, surround fine aqueous pores. Paracellular spaces or
channels also exist between certain epithelial/endothelial cells. Other
adsorbed proteins have enzymatic, carrier, receptor or signal transduction properties.
Lipid molecules also are capable of lateral movement. Thus, biological
membranes are highly dynamic structures.
Drugs are transported
across the membranes by:
Passive diffusion and filtration
Specialized transport
Passive diffusion
The drug diffuses across the membrane in the direction of its concentration gradient, the membrane playing no active role in the process. This is the most important mechanism for majority of drugs; drugs are foreign substances (xenobiotics), and specialized mechanisms are developed by the body primarily for normal metabolites.
Lipid soluble drugs
diffuse by dissolving in the lipoidal matrix of the membrane (Fig. 2.3), the
rate of transport being proportional to the lipid : water partition coefficient
of the drug. A more lipidsoluble drug attains higher concentration in the
membrane and diffuses quickly. Also, greater the difference in the
concentration of the drug on the two sides of the membrane, faster is its diffusion.
Influence of pH Most drugs are weak
electrolytes, i.e. their ionization is pH dependent (contrast strong
electrolytes that are nearly completely ionized at acidic as well as alkaline
pH). The ionization of a weak acid HA is given by the equation:
[A¯ ]
pH = pKa + log —–—
...(1)
[HA]
pKa is the negative
logarithm of acidic dissociation constant of the weak electrolyte. If the
concentration of ionized drug [A¯ ] is equal to concentration of unionized drug
[HA], then
[A¯ ]
—–— = 1
[HA]
Since log 1 is 0, under this condition
pH = pKa ...(2)
Thus, pKa
is numerically equal to the pH at which the drug is 50% ionized.
If pH is increased by 1 scale, then—log [A¯ ]/[HA] = 1 or [A¯ ]/[HA] = 10
Similarly,
if pH is reduced by 1 scale, then— [A¯ ]/[HA] = 1/10
Thus,
weakly acidic drugs, which form salts with cations, e.g. sod. phenobarbitone, sod.
sulfadiazine, pot. penicillinV, etc.
ionize more at alkaline pH and 1 scale change in pH causes 10 fold change in
ionization.
Weakly basic drugs,
which form salts with anions, e.g. atropine sulfate,
ephedrine HCl, chloroquine phosphate, etc. conversely ionize more
at acidic pH. Ions being lipid insoluble, do not diffuse and a pH difference
across a membrane can cause differential distribution of weakly acidic and
weakly basic drugs on the two sides (Fig. 2.4).
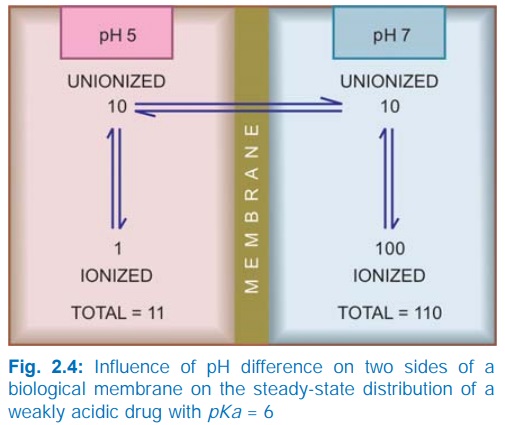
Implications of this
consideration are:
Acidic drugs, e.g. aspirin (pKa
3.5) are largely unionized at acid gastric pH and are absorbed from stomach,
while bases, e.g. atropine (pKa 10)
are largely ionized and are absorbed only when they reach the intestines.
The unionized form of
acidic drugs which crosses the surface membrane of gastric mucosal cell,
reverts to the ionized form within the cell (pH 7.0) and then only slowly
passes to the extracellular fluid. This is called ion trapping, i.e. a weak electrolyte crossing a membrane to
encounter a pH from which it is not able to escape easily. This may contribute
to gastric mucosal cell damage caused by aspirin.
Basic drugs attain higher concentration intracellularly
(pH 7.0 vs 7.4 of plasma).
Acidic drugs are ionized more in alkaline
urine—do not back diffuse in the kidney tubules and are excreted faster.
Accordingly, basic drugs are excreted faster if urine is acidified.
Lipidsoluble
nonelectrolytes (e.g. ethanol, diethylether) readily cross biological membranes
and their transport is pH independent.
Filtration
Filtration is passage
of drugs through aqueous pores in the membrane or through paracellular spaces.
This can be accelerated if hydrodynamic flow of the solvent is occurring under
hydrostatic or osmotic pressure gradient, e.g. across most capillaries
including glomeruli. Lipidinsoluble drugs cross biological membranes by
filtration if their molecular size is smaller than the diameter of the pores
(Fig. 2.3). Majority of cells (intestinal mucosa, RBC, etc.) have very small
pores (4 Å) and drugs with MW > 100 or 200 are not able to penetrate.
However, capillaries (except those in brain) have large paracellular spaces (40
Å) and most drugs (even albumin) can filter through these (Fig. 2.8). As such,
diffusion of drugs across capillaries is dependent on rate of blood flow
through them rather than on lipid solubility of the drug or pH of the medium.
Specialized transport
This
can be carrier mediated or by pinocytosis. Carrier transport
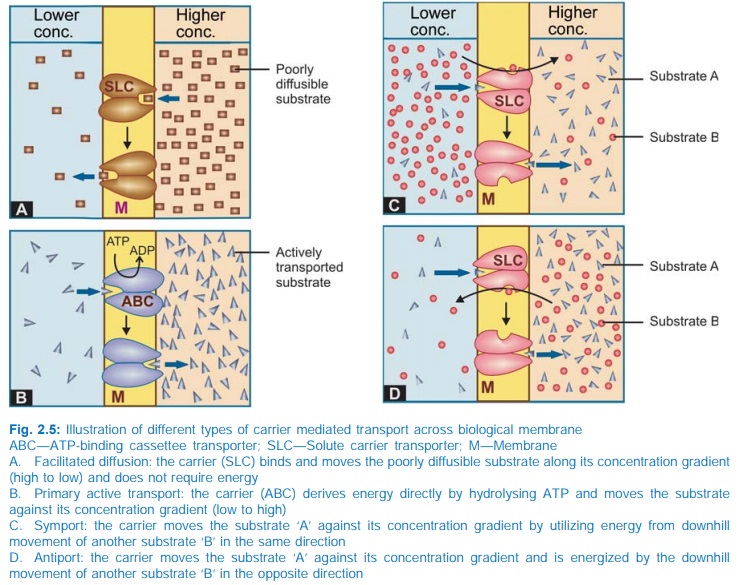
All cell membranes
express a host of transmembrane proteins which serve as carriers or
transporters for physiologically important ions, nutrients, metabolites,
transmitters, etc. across the membrane. At some sites, certain transporters
also translocate xenobiotics, including drugs and their metabolites. In
contrast to channels, which open for a finite time and allow passage of
specific ions, transporters combine transiently with their substrate (ion or
organic compound)—undergo a conformational change carrying the substrate to the
other side of the membrane where the substrate dissociates and the transporter
returns back to its original state (Fig. 2.5). Carrier transport is specific
for the substrate (or the type of substrate, e.g. an organic anion), saturable,
competitively inhibited by analogues which utilize the same transporter, and is
much slower than flux through channels. Depending on requirement of energy,
carrier transport is of two types:
a. Facilitated diffusion The transporter,
belonging to the superfamily of solute
carrier (SLC) transporters, operates passively without needing energy and
translocates the substrate in the direction of its electrochemical gradient,
i.e. from higher to lower concentration (Fig. 2.5A). It mearly facilitates
permeation of a poorly diffusible substrate, e.g. the entry of glucose into
muscle and fat cells by GLUT 4.
b. Active transport It requires energy,
is inhibited by metabolic poisons, and transports the solute against its electrochemical
gradient (low to high), resulting in selective accumulation of the substance on
one side of the membrane. Drugs related to normal metabolites can utilize the
transport processes meant for these, e.g. levodopa and methyl dopa are actively
absorbed from the gut by the aromatic amino acid transporter. In addition, the
body has developed some relatively nonselective transporters, like Pglycoprotein (Pgp), to deal with
xenobiotics. Active transport can be
primary or secondary depending on the source of the driving force.
i)
Primary
active transport
Energy is obtained
directly by the hydrolysis of ATP (Fig. 2.5B). The transporters belong to the
superfamily of ATP binding cassettee (ABC) transporters
whose intracellular loops have ATPase activity. They mediate only efflux of the
solute from the cytoplasm, either to extracellular fluid or into an
intracellular organelli (endoplasmic reticulum, mitochondria, etc.)
Encoded by the
multidrug resistance 1 (MDR1) gene, Pgp is the most well known primary active transporter
expressed in the intestinal mucosa, renal tubules, bile canaliculi, choroidal
epithelium, astrocyte foot processes around brain capillaries (the bloodbrain
barrier), testicular and placental microvessels, which pumps out many
drugs/metabolites and thus limits their intestinal absorption, penetration into
brain, testes and foetal tissues as well as promotes biliary and renal
elimination. Many xenobiotics which induce or inhibit Pgp also have a similar
effect on the drug metabolizing isoenzyme CYP3A4, indicating their synergistic
role in detoxification of xenobiotics.
Other primary active
transporters of pharmacological significance are multidrug resistance
associated protein 2 (MRP 2) and breast cancer resistance protein (BCRP).
ii)
Secondary active transport
In
this type of active transport effected by another set of SLC transporters, the
energy to pump one solute is derived from the downhill movement of another
solute (mostly Na+). When the concentration gradients are such that both the solutes
move in the same direction (Fig. 2.5C), it is called symport or cotransport,
but when they move in opposite directions (Fig. 2.5D), it is termed antiport or exchange transport. Metabolic energy (from hydrolysis of ATP) is spent in maintaining high transmembrane electrochemical
gradient of the second solute. The SLC transporters mediate both uptake and
efflux of drugs and metabolites.
The organic anion
transporting polypeptide (OATP) and organic cation transporter (OCT), highly
expressed in liver canaliculi and renal tubules, are secondary active
transporters important in the metabolism and excretion of drugs and metabolites
(especially glucuronides). The Na+,Cl– dependent neurotransmitter transporters
for serotonin and dopamine (SERT and DAT) as well as the vesicular transporter
for biogenic amines are active SLC transporters that are targets for action of
drugs like tricyclic antidepressants and reserpine, etc. The absorption of
glucose in intestines and renal tubules is through secondary active transport by
sodiumglucose transporters (SGLT1 and SGLT2).
As indicated earlier,
carrier transport (both facilitated diffusion and active transport) is
saturable and follows the Michaelis Menten kinetics. The maximal rate of transport
is dependent on the density of the transporter in a particular membrane, and
its rate constant (Km), i.e. the substrate concentration at which rate of
transport is half maximal, is governed by its affinity for the substrate.
Genetic polymorphism can alter both the density and affinity of the transporter
protein for different substrates and thus affect the pharmacokinetics of drugs.
Moreover, tissue specific drug distribution can occur due to the presence of
specific transporters in certain cells.
Pinocytosis It is the process of transport across the cell in particulate form by formation of vesicles.
This is applicable to proteins and other big molecules, and contributes little
to transport of most drugs.
