Platelet Plug Formation
| Home | | Biochemistry |Chapter: Biochemistry : Blood clotting
Platelets (thrombocytes) are small, anucleate fragments of megakaryocytes that adhere to exposed collagen of damaged endothelium, get activated, and aggregate to form a platelet plug.
PLATELET PLUG FORMATION
Platelets
(thrombocytes) are small, anucleate fragments of megakaryocytes that adhere to
exposed collagen of damaged endothelium, get activated, and aggregate to form a
platelet plug (Figure 34.20). Formation of the platelet plug is referred to as
primary hemostasis. In a normal adult there are 150,000–450,000 platelets per
μl of blood. They have a lifespan of up to 10 days, after which they are taken
up by the liver and spleen and destroyed. Clinical laboratory tests to measure
platelet number and activity are available.
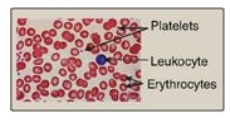
Figure 34.20 Size comparison of platelets, erythrocytes, and a leukocyte.
A. Adhesion
Adhesion of platelets
to exposed collagen at the site of vessel injury is mediated by the protein von
Willebrand factor (VWF). VWF binds to collagen, and platelets bind to VWF via
glycoprotein Ib (GPIb), a component of a membrane receptor complex (GPIb– V–IX)
on the platelet surface (Figure 34.21). Binding to VWF stops the forward
movement of platelets. [Note: Deficiency in the receptor for VWF results in
Bernard-Soulier syndrome, a disorder of decreased platelet function and
number.] VWF is a glycoprotein that is released from platelets. It also is made
and secreted by endothelial cells. In addition to mediating the binding of
platelets to collagen, VWF also binds to and stabilizes FVIII in the blood.
Deficiency of VWF results in von Willebrand disease (VWD), the most common
inherited defect in the ability to clot (coagulopathy). VWD coagulopathy
results from decreased binding of platelets to collagen and a deficiency in
FVIII (due to increased degradation). Platelets can also bind directly to
collagen via the membrane receptor GPVI. Once adhered, platelets get activated.
[Note: Damage to the endothelium also exposes FIII, initiating the extrinsic
pathway of blood clotting and activation of FX (see Figure 34.8).]
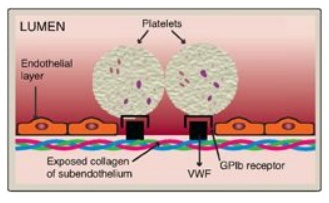
Figure 34.21 Binding of platelets via the receptor glycoprotein Ib (GPIb) to von Willebrand factor (VWF). VWF is bound to the exposed collagen at a site of injury.
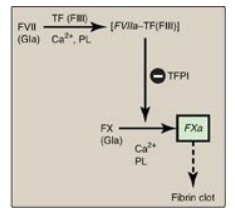
Figure 34.8 The extrinsic or tissue factor (TF) pathway. Binding of FVII to exposed TF (FIII) activates FVII. [Note: The pathway is quickly inhibited by tissue factor pathway inhibitor (TFPI).] F = factor; Gla = γ-carboxyglutamate; PL = phospholipid; a = active.
B. Activation
Once adhered to areas
of injury, platelets get activated. Platelet activation involves morphologic
(shape) changes and degranulation, the process by which platelets secrete the
contents of their α and δ (or dense) storage granules. Activated platelets also
expose PS on their surface. Thrombin is the most potent platelet activator.
Thrombin binds to and activates protease-activated receptors, a type of G
protein– coupled receptor (GPCR), on the surface of platelets (Figure 34.22) .
Thrombin is primarily associated with Gq proteins, resulting in activation of
phospholipase C and a rise in diacylglycerol (DAG) and inositol trisphosphate
(IP3). [Note: Thrombomodulin, through its binding of thrombin, decreases the
availability of thrombin for platelet activation (see Figure 34.18).]
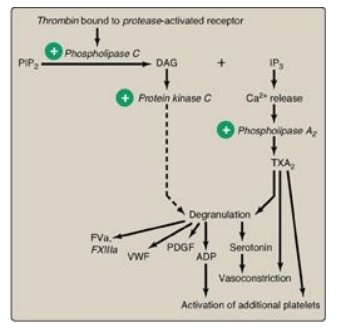
Figure 34.22 Platelet activation by thrombin. [Note: Protease-activated receptors are a type of G protein-coupled receptor.] PIP2 = phosphoinositol bisphosphate; DAG = diacylglycerol; IP3 = inositol trisphosphate;TXA2 = thromboxane A2; ADP = adenosine diphosphate; PDGF = platelet-derived growth factor; VWF = von Willebrand factor; F = factor.
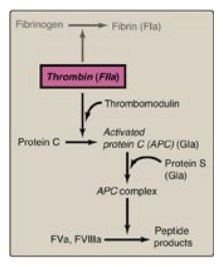
Figure 34.18 Formation and action of the APC complex. Gla = γ-carboxyglutamate; a = active; F = factor.
1. Degranulation: DAG activates protein kinase C, a key event for
degranulation. IP3 causes the release of Ca2+ (from dense
granules). The Ca2+ activates phospholipase A2, which
cleaves membrane phospholipids to release arachidonic acid, the substrate for
the synthesis of thromboxane A2 (TXA2) in activated
platelets by cyclooxygenase-1 (COX-1). TXA2 causes vasoconstriction,
augments degranulation, and binds to platelet GPCRs, causing activation of
additional platelets. Recall that aspirin irreversibly inhibits COX and,
consequently, TXA2 synthesis and is referred to as an “antiplatelet”
drug. Degranulation also results in release of serotonin and adenosine
diphosphate (ADP) from dense granules. Serotonin causes vasoconstriction. ADP
binds to GPCRs on the surface of platelets, activating additional platelets.
[Note: Some antiplatelet drugs, such as clopidogrel, are ADP-receptor
antagonists.] Platelet-derived growth factor (involved in wound healing), VWF,
FV, FXIII, and fibrinogen are among other proteins released from α granules. [Note:
Platelet-activating factor (PAF), an ether phospholipid synthesized by a
variety of cell types including endothelial cells and platelets, binds PAF
receptors (GPCRs) on the surface of platelets and activates them.]
2. Shape change: The change in shape of activated platelets from
discoidal to spherical with pseudopod-like processes that facilitate platelet–platelet
and platelet–surface interactions (Figure 34.23) is initiated by the release of
Ca2+ from dense granules. Ca2+ bound to calmodulin mediates
the activation of myosin light-chain kinase that phosphorylates the myosin
light chain, resulting in a major reorganization of the platelet cytoskeleton.
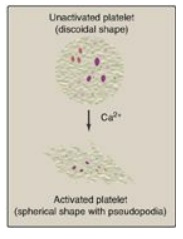
Figure 34.23 Activated
platelets undergo Ca2+-initiated shape change.
C. Aggregation
Activation causes
dramatic changes in platelets that lead to their aggregation. Structural
changes in a surface receptor (GPIIb/IIIa) expose binding sites for fibrinogen.
Bound fibrinogen molecules link activated platelets to one another (Figure 34.24),
with a single fibrinogen able to bind two platelets. The fibrinogen is
converted to fibrin by thrombin and then covalently cross-linked by FXIIIa
coming from both the blood and the platelets. [Note: The exposure of PS on the
surface of activated platelets allows formation of the Xase complex (VIIIa,
IXa, X, and Ca2+) with subsequent formation of FXa and generation of
thrombin.] Fibrin strengthens the platelet plug. [Note: Rare defects in the
platelet receptor for fibrinogen result in Glanzmann thrombasthenia (decreased
platelet function), whereas autoantibodies to this receptor are a cause of
immune thrombocytopenia (decreased platelet number).]
Unnecessary activation of platelets is prevented
because 1) an intact vascular wall is separated from the blood by a monolayer
of endothelial cells, preventing the contact of platelets with collagen; 2)
endothelial cells synthesize prostaglandin I2 (PGI2, or
prostacyclin) and nitric oxide, each of which causes vasodilation; and 3)
endothelial cells have a cell-surface ADPase that converts ADP to AMP.
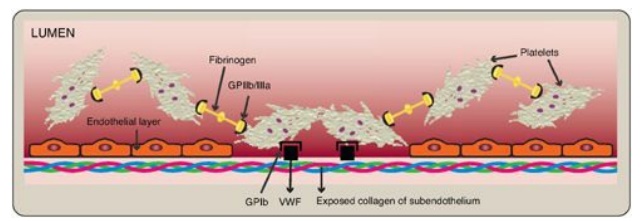
Figure 34.24 Linking of
platelets by fibrinogen via the receptor glycoprotein (GP) IIb/IIIa. [Note: The
shapes in the fibrinogen molecule represent the two D and one E domains.] GPIb
= glycoprotein Ib receptor; VWF = von Willebrand factor.
Related Topics
