Process Analytical Technology
| Home | | Pharmaceutical Technology |Chapter: Pharmaceutical Engineering: Process Analytical Technology
To maximize the control over any process in pharmaceutical product development, information is required on the way in which the product is responding to changes in manufacturing variables.
Process Analytical
Technology
To
maximize the control over any process in pharmaceutical product development,
information is required on the way in which the product is responding to
changes in manufacturing variables. Historically, the information was derived
from data obtained on the nature of batches produced under particular
manu-facturing conditions. Knowledge of the batch properties were employed to
modify the manufacturing conditions to ensure that the product was closely
controlled to designated quality specifications.
In
recent years, analytical methods and their application have improved to the
point that real time in process measurements can be taken and fed back through
control systems to the input parameters to allow for continuous mon-itoring and
control of processes.
In
earlier chapters, examples of the major unit operations in pharmaceu-tical
manufacturing were outlined. These processes will now be considered with
anecdotal evidence from the literature of methods that might lead to closer
control of the product quality and thereby conform to recent regulatory
direc-tives to consider such methods as part of the Quality by Design (QbD)
initiative.
The
Food and Drug Administration has issued a guidance document on Process
Analytical Technology (PAT) (Zu et al., 2007). Processes may be divided into
batch and continuous approaches. These processes can be monitored by in situ,
real time, and/or feedback control analyses to assure the quality of the
product (Fig. 19.1).
PAT
necessarily begins with the manufacture of active pharmaceutical ingredient
(API) and any additives and understanding their properties (Byrn et al., 2006;
Hlinak et al., 2006). Important methods in this context address the presence of
impurities (including moisture), degradation products (stability), component
compatibility, and crystallinity (polymorphism). Near infrared spectroscopy has
been applied in situ, real time to address the chemical com-position of API or
additive during manufacture (Mendendorp et al., 2006). Near infrared laser
Raman spectroscopy has been employed to monitor polymeriza-tion process
(Francisco et al., 2006). A variety of particle sizing methods can be employed,
but those that are in situ, real time employ laser scattering methods. Dosage
form manufacturing can be optimized by direct methods of monitoring the
variables involved in drying, mixing/blending (Portillo, et al., 2008),
gran-ulation (Papp et al., 2008), filling, compression (Askeli and Cetinkaya,
2008; Soh et al., 2007), and coating (Bose et al., 2006; Cogdill et al., 2007).
For more sophisticated dosage forms, compatibility with packaging components is
also required, but this is likely to have been considered during the
preliminary experimental design optimization steps.
PAT
arguably is at the intersection of design space (considered in chap. 18) and
control strategy, these being the major elements of QbD. These topics have been
described in Product Quality Lifecycle Implementation initiative of the
International Society for Pharmaceutical Engineering (Drennan, 2008). Topics of
interest in this initiative have been described in the Journal of
Pharmaceutical Innovation.
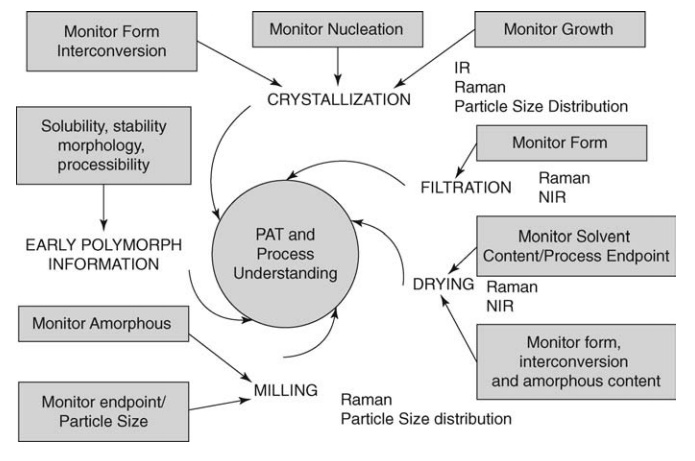
FIGURE 19.1
Critical steps in API manufacture. Source: Modified from Byrn et al. (2006).
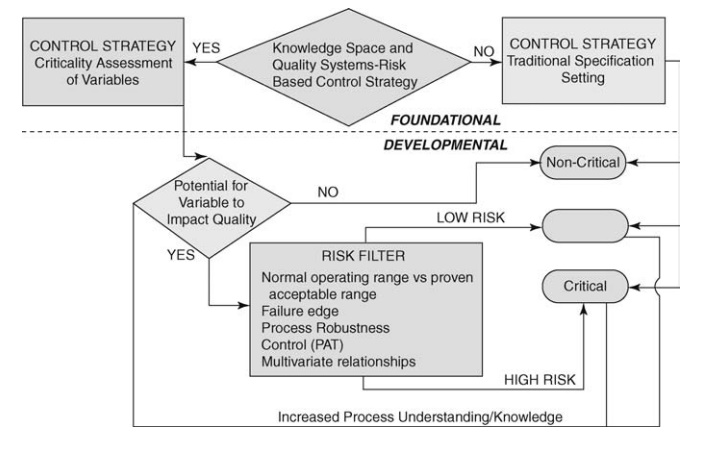
FIGURE 19.2 Decision tree to define levels of criticality. Source: Modified from
Garcia et al. (2008).
Figure
19.2 presents the most prominent decisions required to evaluate the criticality
of variables in process development. Decisions (diamonds) are made based on the
business decision in foundational classification (above dotted line), and risk
assessment in developmental classification (below dotted line) that pass
through a filter (rectangle) to criticality designations (rounded rectangle).
With respect to criticality, those variables that are not critical have not
been demonstrated to impact on safety or efficacy or factor into critical
quality attributes (CQA) as defined by ICH Q(8) R and consequently do not have
to be included in design space. Critical variables are those that are known to
impact safety, efficacy, or other measures of biological disposition or
com-pliance. Critical process parameters if varied beyond a certain range have
a direct and significant influence on CQAs. These properties must be controlled
within predesignated range to ensure final product quality. The empty symbol
represents an alternative designation for attributes that may impact the
product but represent a low risk. The designation of low risk is based on an
indirect impact on safety and/or efficacy alone or in combination with other
variables; mitigated risk; and knowledge transfer from noncritical variables
requiring additional evaluation.
It
has been suggested that criticality can be reduced to fundamental ele-ments of
severity, occurrence, and detection in a compounding manner (Nosal and Schultz,
2008). These terms can be related to experimental design (frequency and
variation) and analytical capability (detection). During the life cycle of the
product clear differentiation of levels of criticality is required to address a
control strategy based on process variables, material attributes, and their
relationship to quality measures.
Related Topics
