Quality Control and Quality Assurance
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Sterile Pharmaceutical Products
It is not the aim of this section to review the entirety of quality control of sterile products or of the chemical assays and requirements of drugs and excipients prior to formulation. Only those techniques with importance either to microbiology or the confirmation of sterility of the final product are introduced here.
QUALITY CONTROL AND QUALITY
ASSURANCE
It is not the
aim of this
section to review
the entirety of quality control of sterile
products or of the chemical assays and requirements of drugs and excipients prior to
formulation. Only those
techniques with importance either to microbiology or the confirmation of sterility of the final product
are introduced here.
A) Bioburden
It should
be obvious from
previous sections that a successful sterilization process is dependent on a product having a low presterilization bioburden. This will
also be true of the individual ingredients, which must
have low levels of microbial contamination, or else there is a
danger that
the contaminants will
find their way into the final product or be a source
of pyrogens.
Sterilization should
normally be considered as the removal of the bioburden, but the high heat resistance of bacterial endotoxins means that successful steam sterilization does not necessarily guarantee
that the product will pass a pharmacopoeial endotoxin
test; dead bacteria are likely to remain pyrogenic.
Underestimating the
level of microbial contamination prior to the terminal sterilization process will lead to a miscalculation of the sterilization dose requirements to achieve the desired SAL.
The bioburden must
be maintained within certain
limits to justify
the chosen sterilization process. When a higher
number of organisms or more
resistant microorganisms are encountered during manufacture of batches than was determined during the initial validation, those
batches must be assumed not to
be sterile. The bioburden is an estimate
of the total
viable count of microorganisms present
before sterilization, and a knowledge of the resistance characteristics
of these organisms is often an integral part
of the sterility assurance calculation. To build
some degree of safety into
the sterilization process
the sterilization conditions should be set to destroy
all the bioburden by assuming that all the
contaminating
microorganisms are the most resistant of the species identified in that bioburden. Sterility assurance, as implied
in the schemes shown in Figures 22.1 and
22.2, can only be achieved with
a low bioburden and with fully validated, correctly functioning sterilizers.
B) The Test For Sterility
The broad
basis of the test for sterility is that it examines
samples of the final product
for the presence of microorganisms.
Theoretically, the test for sterility should
be applied to all products that are designated as sterile. However, the test does not examine
all samples in a batch, and its results
can only be considered valid
if all items in a batch
are treated similarly (British
Pharmacopoeia, 2010). Clearly,
for products which are terminally sterilized this might
seem a reasonable assumption but only if
there is uniform
heat distribution in an autoclave
or hot air oven
or uniform delivery
of a radiation dose. With
aseptically produced
products there are dangers because not all items
in a batch may have been treated
similarly. A successful test
only shows that
no microbial contamination was found in the samples
examined under the test
conditions. Extension of the result
to a whole batch
requires the assurance that every
unit in the
batch was manufactured in such a manner that it would
also have passed the test with a high degree of probability. This highlights the weakness of the test
for sterility and why
the controls
of sterilization processes
are very important and probably of greater assurance in confirming the
sterility of a batch.
The test, however, remains
one of few analytical methods that examine a product for sterility.
C) Parametric Release
As there are significant limitations with the test for sterility, many authorities place
considerable reliance on the
validation and reliable performance of sterilizers and their sterilization cycles.
Parametric release takes
this reliance a step further by allowing batches
of terminally sterilized products to be released
without being subjected to the test
for sterility. The sterilization cycle
will be validated to have a SAL of 10−6 or less as the minimum
safety factor. Validation studies would include
heat distribution, heat penetration, bioburden, container closure
and cycle lethality studies.
For a product to be subject to parametric
release, presterilization bioburden testing on each batch
would be completed, and the comparative resistance of isolated spore-formers checked. Each cycle would
include the use of chemical
or biological indicators. It is hoped that these actions will provide a significantly higher
level of assurance of sterility than provided by the test for
sterility. This
requires confirmation that
each part of the
manufacturing process
has been satisfactorily completed, the initial presterilization bioburden is within agreed limits, that
the controls for the sterilizing cycle were satisfactory and that the correct time
cycles were achieved. In practice parametric release
should only be used when experience has been gained
on a reliably controlled and adequately validated process
and where a relationship has been
proved between end-product testing and in-process monitoring.
Clearly,
reproducibility,
regular monitoring and documentation are required. However, parametric
release would imply abandoning the sterility test,
an option that many manufacturers have not yet
adopted, possibly because of the fear of litigation based
on the premise that any sterile
product would, if tested, have
passed the test for sterility.
D) Pyrogens
The discovery that
aqueous solutions may lead to an increase in body temperature when injected into a patient dates back to the 19th century. The agents responsible for this fever
were termed ‘pyrogens’. In theory a pyrogen is any substance that, when injected
into a mammal, elicits a rise in body temperature, and substances produced
by some Gram-positive bacteria, mycobacteria, fungi and also viruses conform
to this definition. The most common
pyrogens, however, and those of major significance to the
pharmaceutical industry, are produced by Gram-negative
bacteria and are known
as endotoxins; they are lipopolysaccharides (LPS)
found in the
cell envelope . The
presence of pyrogens in aqueous solutions was first
demonstrated by injection into rabbits whose
body temperature was recorded. More sensitive methods have since been developed, mostly based on the discovery that a fraction of the horseshoe crab blood reacts
with LPS as a
clotting agent.
Two pharmacopoeial
limit tests exist.
That for pyrogens uses rabbits to assess pharmacological
activity and therefore the presence of pyrogens of all kinds.
The test for bacterial endotoxins uses lysed
amoebocytes (blood cells) of the horseshoe crab and is therefore termed
the Limulus amoebocyte lysate (LAL) test. This may be extended to many drug and device products
and clearly will be developed in the future
to assess the
presence of endotoxins in biotechnology products.
i) Physiological effects of pyrogens
The most characteristic
effect following injection of pyrogens into humans
is a rise of body
temperature, but it is only one of a number
of dose-dependent diverse effects. Pyrogens elevate
the circulating levels
of inflammatory cytokines, which may be followed by fever,
blood coagulation,
hypotension, lymphopenia, neutrophilia, elevated levels of plasma cortisol and acute-phase proteins. Low doses of pyrogens induce asymptomatic inflammatory reactions. Moderate doses induce
fever and changes in plasma composition. Injection of high pyrogenic doses results in shock, characterized by cardiovascular dysfunction, vasodilation,
vasoconstriction, endothelium dysfunction and multiple
organ dysfunction or failure and death.
ii) Characteristics of bacterial endotoxin
The release of LPS from
bacteria takes place
after death and lysis. Many
Gram-negative bacteria, e.g. Escherichia coli and Proteus,
Pseudomonas, Enterobacter and Klebsiella species produce pyrogenic LPS which is composed of two
main parts: a hydrophilic polysaccharide chain with antigenic regions, and
a hydrophobic lipid
group termed lipid A which is responsible for many of the biological activities. The molecular
size of the polysaccharide chain is very variable, and consequently the molecular weight of the LPS may vary from a few thousand
to several million daltons. LPS
is unusually thermostable and in sensitive to pH changes. Molecules are able to withstand 120 °C for over
3 hours. Extremes of pH are required for rapid destruction of the LPS.
iii)
Sources
The sources
of pyrogens in parenteral products include water used at the end
stages of the purification and
crystallization of the drug or excipients; water
used during processing; packaging components; and the
chemicals, raw materials
or equipment used in the preparation of the
product. The presence of endotoxins on devices may be
attributed to water
in the manufacturing process, the washing of components such as filter
media to be used
for the manufacture of filters, or the washing/rinsing of tubing or other
plastic devices prior
to their sterilization. Additionally, if the drug is biologically produced, incomplete removal of the microorganisms during purification
can result
in high endotoxin levels.
iv)
Measurement of pyrogens
Pyrogens have
traditionally been assessed using rabbits which are stored in carefully
controlled conditions and whose
temperature is monitored before the administration of the test product.
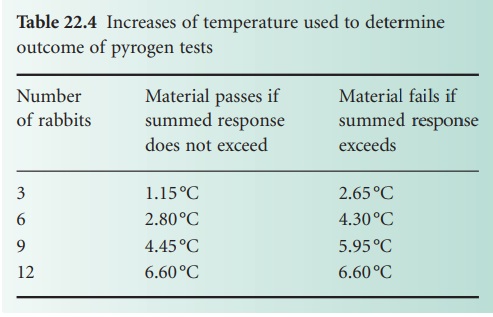
The British Pharmacopoeia (2010) describes a test initially
based on three rabbits;
the number is progressively increased if the results
fall between the two values (Table 22.4). Samples
of the product under test are injected
into the marginal
ear vein at a dose no greater than
10 ml/kg. The animals are monitored for the 3 hour period immediately after injection, at 30 minute intervals. The test assumes that
the maximum rise in temperature will be detected
in this 3 hour
period immediately after injection. Table 22.4 describes the criteria for pass or fail as the number
of rabbits used increases to the maximum
of 12.
A number
of limitations of the rabbit
pyrogen test are recognized. Repeated use of animals leads
to endotoxin tolerance. There is low reactivity to the endotoxin produced by certain
species, e.g. Legionella. There
is also variability
in control results
when identical standardized endotoxin preparations are used, which is
probably related to interlaboratory factors and variations due to seasons, rabbit species
and other biological sources. Care must be taken in testing radiopharmaceuticals,
and certain drugs
may themselves elicit
a rise in temperature on administration. The test is therefore inadequate for radiopharmaceuticals,
cancer chemotherapeutic agents, hypnotics and narcotics, vitamins, steroids and some
antibiotics. The presence of pyrogens may be hidden
by the pharmacological activity of the product’s components. Finally the rabbit test is insufficiently sensitive to detect endotoxin in intrathecal products
where only low levels of pyrogens
are acceptable.
EMA (2009)
is to encourage the replacement of the rabbit test with
the monocyte activation test (Hoffmann,
2005) for plasma-derived medicinal products.
Human monocytes from cultured
cell lines mimic the human fever reaction in vitro
by producing cytokines. Cytokine release can be determined, usually using enzyme-linked immunoassay (ELISA).
v)
Measurement of bacterial endotoxins
The LAL test is considerably more
sensitive than the pyrogen test. As mentioned above,
although the Legionella
endotoxin is not very pyrogenic to rabbits it is easily detected by the LAL test.
It has been
estimated that there is a 1000-fold difference in sensitivity between
the two tests, but the LAL test only
detects endotoxins of Gramnegative bacteria
and not all pyrogens. However, the LAL
test may be used
for radiopharmaceuticals.
LAL test reagent
comes from the American horseshoe crab Limulus
polyphemus. The endotoxin-induced coagulation of its blood is based on an enzyme-mediated interaction
of LAL with endotoxins. The reagents are obtained from the blood of freshly captured
horseshoe crabs whose
amoebocytes are concentrated, washed
and lysed with endotoxin-free water. The LAL is separated
from the remaining cellular debris and its activity optimized
using metallic cations, pH adjustment and
additives and then
freeze-dried. Certain preparations interfere with the interaction between LAL and endotoxin. Chemical
inhibitors may cause chelation of the
divalent cations necessary for the reaction, protein denaturation
or inappropriate pH changes. Physical inhibition may result from adsorption of endotoxin or be caused by viscosity of the product.
Even the type of glassware may affect the test. Siliconized glassware or plastic can
inhibit gel-clot formation, or prevent accurate spectrophotometric readings of the reaction
end-point.
The samples
of products are incubated with LAL at 37 °C. If endotoxins are
present a solid
gel forms, indicating the presence
of endotoxins. The British Pharmacopoeia
(2010) describes six
separate methodologies for
the test for endotoxin. These
are (A) gel-clot
limit test; (B) gelclot: semi quantitative; (C) turbidimetric
kinetic method; (D) chromogenic
kinetic method; (E)
chromogenic endpoint method;
and (F) turbidimetric end-point method.
There are checks for interfering factors. Any validated
method may be used, but the gel-clot
method is the referee test in the case of dispute. Coloured products cannot be tested
by turbidimetric and chromogenic
methods, as precipitate formation may be mistaken for a
positive response.
Kinetic LAL methods are claimed to increase the efficiency of large-scale testing,
probably important when validation of depyrogenation cycles
or preparation of components for aseptic
processing are required. For all procedures, test validation must be conducted to rule out interference, which may be either
inhibition or enhancement. Depyrogenated glassware must be
used throughout.
The gel-clot method
is most commonly used. The test is conducted by adding the LAL reagent
to an equal volume of test
solution, agitating and storing at 37 °C for
1 hour when
the end-point is determined by inversion
of the tubes. If a solid clot remains intact,
the product is considered to contain endotoxins.
Chromogenic methods utilize
colorimetry but do not depend
on the clottable protein. A synthetic substrate
is used that contains an amino
acid sequence similar
to that of coagulogen, the clottable
protein. The activated
proclotting enzyme cleaves
a p-nitroanilide chromophore from the
synthetic substrate and the colour
produced is proportional to the amount of endotoxin. The turbidimetric
LAL method is based on the fact
that an increase in endotoxin concentration will cause a proportional increase
in turbidity caused
by the precipitation of the clottable protein, coagulogen. The optical density
is read spectrophotometrically either at a fixed time or constantly for kinetic assays
as turbidity develops. The kinetic methods depend on the
relationship between the logarithm of the
response and the logarithm of the endotoxin concentration. The end-point
methods relate endotoxin
levels to the quantity
of chromophore released
or the amount
of precipitation.
vi)
Endotoxins in
parenteral Pharmaceuticals
The limits
for endotoxin are based on the dose
of the product. Put simply, the endotoxin limit,
EL, which represents the maximum amount of endotoxin that is allowed in a specific dose,
is inversely related
to the dose of
the drug; it may be assessed from
the following equation (United States Pharmacopeia, 2010):
EL = K/M (2)
where K is
the threshold human
pyrogenic dose of endotoxin per kg body
weight and M is
the maximum human dose of the product in kg body weight that would be administered
in a single 1 hour
period. M recognizes that the pharmacological effects of endotoxin are
dosedependent. The endotoxin limit is the
level at which
a product is
adjudged pyrogenic or non-pyrogenic. Gelclot reagent sensitivities are generally in the range
0.015–0.5 EU/ml. As examples of endotoxin limits,
the United States Pharmacopeia (2010) states limits
of no more than 0.5 EU/ml for Dextrose Infusion, no more than 5 EU/mg promethazine in Promethazine Injection
USP, no more than 10 EU/mg of mitomycin in Mitomycin for
Injection USP and no more than 24 EU/mg warfarin
sodium in Warfarin Sodium for Injection. The British Pharmacopoeia
(2010) has a limit
of 0.25 IU/ml
in Glucose Intravenous Infusion; this
value is similar
for many BP intravenous
infusions. As another example,
insulin should contain
no more than 10 IU/mg
of endotoxin. The endotoxin limit for drugs gaining
access to the cerebrospinal fluid
is reduced to 0.2 EU/kg
because the intrathecal route is the most toxic route for endotoxins.
vii)
Depyrogenation and the
Production of Apyrogenic Products
Pyrogens and endotoxins
are difficult to remove from products once present
and it is easier to keep components relatively endotoxin-free rather than to remove them from the final product.
Rinsing or dilution is one way of
eliminating pyrogenic activity provided that the rinsing
fluid is apyrogenic. Closures
and vials should be washed with pyrogen-free water
before sterilization. Pyrogens in vials or glass
components may be destroyed by dry heat sterilization at high temperatures. A
recommended condition for depyrogenation of glassware and equipment is heating at 250 °C for 45 minutes.
Pyrogens are also destroyed at 650 °C in 1 minute
or at 180 °C in 4 hours. The British Pharmacopoeia (2010) states that dry heat at
temperatures above 220
°C may be used for
the depyrogenation of glassware. Sterilizing tunnels are designed
not only to sterilize at 250–300 °C but also
to remove pyrogens. These processes equate to incineration, although removal by
washing, also termed dilution, may be used. Filtration, irradiation or ethylene oxide treatment have limited
value in reducing pyrogen or endotoxin loads.
The removal
of pyrogens from
Water for Injections may be effected by distillation or reverse osmosis. Distillation is the most reliable method for removing endotoxin. Care
has to be taken to avoid splashing in the still as pyrogens have been carried
over in droplets. Another source
of endotoxins is the Water for Injection system. Generally, circulating hot water at temperatures above 75 °C provides
an environment that is
not conducive to microbial growth
and thus the formation
of endotoxin. Circulating water at approximately 60 °C causes some concern
as some Gram-negative organisms, e.g. Legionella
pneumophila, will survive and grow at 57 °C. The water-producing systems
may be sanitized by circulating water at 75–80
°C.
Pyrogen-free water
can be produced using an ultrafiltration membrane with a nominal
molecular weight limit that
is low enough to ensure the removal of endotoxins under all conditions. Hollow
fibre polysulphone membranes can be sanitized
with sodium hydroxide, which efficiently destroys pyrogens. A nominal
molecular weight limit of 5000 Da should efficiently
remove endotoxins. However, many endotoxin-producing
microorganisms multiply in ambient temperature Water for Injection systems, especially reverse osmosis (RO) systems, in which the filters
are not absolute
and may be used in series in order to manufacture pyrogenfree water.
Related Topics
