Receptors
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacodynamics Mechanism Of Drug Action; Receptor Pharmacology
The largest number of drugs do not bind directly to the effectors, viz. enzymes, channels, transporters, structural proteins, template biomolecules, etc. but act through specific regulatory macromolecules which control the above listed effectors. These regulatory macromolecules or the sites on them which bind and interact with the drug are called ‘receptors’.
RECEPTORS
The
largest number of drugs do not bind directly to the effectors, viz. enzymes, channels, transporters,
structural proteins, template biomolecules, etc. but act through specific
regulatory macromolecules which control the above listed effectors. These
regulatory macromolecules or the sites on them which bind and interact with the
drug are called ‘receptors’.
Receptor:
It
is defined as a macromolecule or binding site located
on the surface or inside the effector cell that serves to recognize the signal
molecule/drug and initiate the response to it, but itself has no other
function.
Though, in a broad
sense all types of target biomolecules,
including the effectors (enzymes, channels, transporters, etc.) with which a
drug can bind to produce its action have been denoted as ‘receptors’ by some
authors, such designation tends to steal the specific meaning of this important
term. If so applied, xanthine oxidase would be the ‘receptor’ for allopurinol,
Ltype Ca2+ channel would be the ‘receptor’ for nifedipine, serotonin
transporter (SERT) would be the ‘receptor’ for fluoxetine; a connotation not in
consonence with the general understanding of the term. It is therefore better
to reserve the term ‘receptor’ for purely regulatory macromolecules which
combine with and mediate the action of signal molecules including drugs.
The following terms are used in describing drugreceptor
interaction:
Agonist
An agent which
activates a receptor to produce an effect
similar to that of the physiological signal molecule.
Inverse Agonist
An agent which
activates a receptor to produce an
effect in the opposite direction to that of the agonist.
Antagonist
An
agent which prevents the action of an agonist on a
receptor or the subsequent response, but does not have any effect of its own.
Partial agonist
An
agent which activates a receptor to produce
submaximal effect but antagonizes the action of a full agonist.
Ligand
(Latin: ligare—to bind) Any molecule which attaches selectively to particular
receptors or sites. The term only indicates affinity or binding without regard
to functional change: agonists and competitive antagonists are both ligands of
the same receptor.
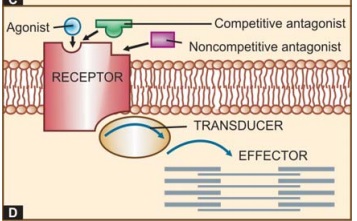
The
overall scheme of drug action through receptors is depicted in Fig. 4.1D.
Basic Evidences For Drug Action Through Receptors
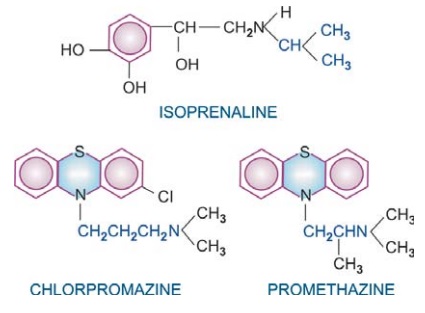
Many drugs exhibit structural specificity of
action, i.e. specific chemical configuration is associated with a particular
action, e.g. isopropyl substitution on the ethylamine side chain of sympathetic
drugs produces compounds with marked cardiac and bronchial activity—most β adrenergic agonists
and antagonists have this substitution. A 3 carbon inter nitrogen separation in
the side chain of phenothiazines results in anti dopaminergic antipsychotic
compounds, whereas 2 carbon separation produces anti-cholinergic antihistaminic
compounds. Further, chiral drugs show stereospecificity in action, e.g. levo noradrenaline is 10 times more
potent than dextro noradrenaline; d-propranolol is about 100 times less
potent in blocking β receptors than the l-isomer,
but both are equipotent local anaesthetics.
Thus, the cell must
have some mechanism to recognize a particular chemical configuration and three
dimensional structure.
Competitive antagonism is seen between specific agonists and
antagonists. Langley in 1878 was so impressed by the mutual antagonism among
two alkaloids pilocarpine and atropine that he proposed that both reacted with
the same ‘receptive substance’ on the cell. Ehrlich (1900) observed
quantitative neutralization between toxins and antitoxins and designated
‘receptor’ to be the anchoring group of the protoplasmic molecule for the
administered compound.
It was calculated by Clark that adrenaline and acetylcholine
produce their maximal effect on frog’s heart by occupying only 1/6000th of the
cardiac cell surface— thus, special regions of reactivity to such drugs must be
present on the cell.
Receptor Occupation Theory
After studying
quantitative aspects of drug action, Clark (1937) propounded a theory of drug
action based on occupation of receptors by specific drugs and that the pace of
a cellular function can be altered by interaction of these receptors with drugs
which, in fact, are small molecular ligands. He perceived the interaction
between the two molecular species, viz.
drug (D ) and receptor (R) to be governed by the law of mass
action, and the effect (E) to be a
direct function of the drug receptor complex (DR) formed:
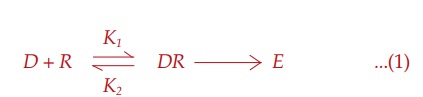
Subsequently, it has
been realized that occupation of the receptor is essential but not itself
sufficient to elicit a response; the agonist must also be able to activate
(induce a conformational change in) the receptor. The ability to bind with the
receptor designated as affinity, and
the capacity to induce a functional change in the receptor designated as intrinsic activity (IA) or efficacy are independent properties. Competitive antagonists
occupy the receptor but do not activate it. Moreover, certain drugs are partial
agonists which occupy and sub-maximally activate the receptor. An all or none
action is not a must at the receptor. A theoretical quantity(S) denoting
strength of stimulus imparted to the cell was interposed in the Clark’s
equation:

Depending
on the agonist, DR could generate a stronger or weaker S, probably as a
function of the conformational change brought about by the agonist in the
receptor. Accordingly:
Agonists have both affinity and maximal intrinsic
activity (IA = 1), e.g. adrenaline, histamine, morphine.
Competitive Antagonists have affinity but no intrinsic activity (IA = 0), e.g. propranolol,
atropine, chlorpheniramine, naloxone.
Partial Agonists have affinity and
submaximal intrinsic activity (IA
between 0 and 1), e.g. di chloro iso-proterenol (on β adrenergic receptor),
pentazocine (on μ opioid receptor).
Inverse Agonists have affinity but
intrinsic activity with a minus sign (IA between 0 and –1), e.g. DMCM (on
benzodiazepine receptor).
It
has also been demonstrated that many full agonists can produce maximal response
even while occupying <1% of the available receptors.
A large receptor
reserve exists in their case, or a number of spare receptors are present.
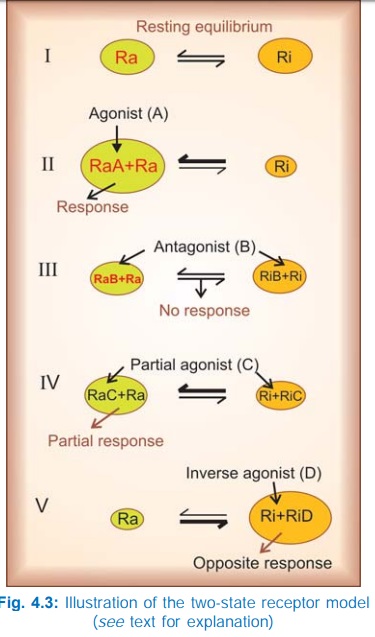
The Two-State Receptor Model
A very attractive
alternative model for explaining the action of agonists, antagonists, partial
agonists and inverse agonists has been proposed.
The receptor is
believed to exist in two interchangeable states: Ra (active) and Ri
(inactive) which are in equilibrium. In the case of majority of receptors, the Ri state is favoured at equilibrium—no/very
weak signal is generated in the absence of the agonist—the receptor exhibits no
constitutive activation (Fig. 4.3I). The agonist (A) binds preferentially to
the Ra conformation and shifts the
equilibrium → Ra predominates and a response is generated (Fig. 4.3II) depending
on the concentration of A. The competitive antagonist (B) binds to Ra and Ri with equal affinity → the equilibrium is not altered → no response is
generated (Fig. 4.3 III), and when the agonist is applied fewer Ra are available to bind it— response to
agonist is decreased. If an agonist has only slightly greater affinity for Ra than for Ri, the equilibrium is only modestly shifted towards Ra (Fig. 4.3 IV)
even at saturating concentrations → a submaximal response is produced and the
drug is called a partial agonist (C). The inverse agonist (D) has high affinity
for the Ri state (Fig. 4.3V),
therefore it can produce an opposite response, provided the resting equilibrium
was in favour of the Ra state.
Certain receptors (mainly Gprotein coupled ones) such as benzodiazepine,
histamine H2, angiotensin AT1, adrenergic β1 and cannabinoid
receptors exhibit constitutive activation, i.e. an appreciable intensity signal
is generated even in the basal state (no agonist present). In their case the
inverse agonist stabilizes the receptor in the inactive conformation resulting
in an opposite response. Only few inverse agonists are known at present, but as
more receptors with constitutive activation are found, more inverse agonists
are likely to be discovered.
This
model has gained wide acceptance because it provides an explanation for the
phenomenon of positive cooperativity often seen with neurotransmitters, and is
supported by studies of conformational mutants of the receptor with altered
equilibrium.
Nature Of Receptors
Receptors are regulatory
macromolecules, mostly proteins, though nucleic acids may also serve as
receptors. They are no longer hypothetical. Hundreds of receptor proteins have
been isolated, purified, cloned and their primary amino acid sequence has been
worked out. Molecular cloning has also helped in obtaining the receptor protein
in larger quantity to study its structure and properties, and in subclassifying
receptors. The cell surface receptors with their coupling and effector proteins
are considered to be floating in a sea of membrane lipids; the folding,
orientation and topography of the system being determined by interactions
between the lipophilic and hydrophilic domains of the peptide chains with
solvent molecules (water on one side and lipids on the other). Nonpolar
portions of the AA chain tend to bury within the membrane, while polar groups
tend to come out in the aqueous medium. In such a delicately balanced system,
it is not difficult to visualize that a small molecular ligand binding to one site
in the receptor molecule could be capable of tripping the balance (by altering
distribution of charges, etc.) and bringing about conformational changes at
distant sites. Each of the four major families of receptors (described later)
have a well defined common structural motif, while the individual receptors
differ in the details of amino acid sequencing, length of intra/extracellular
loops, etc. Majority of receptor molecules are made up of several nonidentical
subunits (hetero-polymeric), and agonist binding has been shown to bring about
changes in their quaternary structure or relative alignment of the subunits,
e.g. on activation the subunits of nicotinic receptor move apart opening a centrally
located cation channel.
Radioligand binding studies have helped in characterizing and
classifying receptors even when they have been dissociated from the effector
system.
Many drugs act upon physiological
receptors which mediate responses to transmitters, hormones, autacoids and
other endogenous signal molecules; examples are cholinergic, adrenergic,
histaminergic, steroid, leukotriene, insulin and other receptors. In addition,
now some truly drug receptors have been described for which
there are no known physiological
ligands, e.g. benzodiazepine receptor, sulfonylurea receptor, cannabinoid
receptor.
Receptor Subtypes
The delineation of multiple types and subtypes of receptors for
signal molecules has played an important role in the development of a number of
targeted and more selective drugs. Even at an early stage of evolution of
receptor pharmacology, it was observed that actions of acetylcholine could be
grouped into ‘muscarinic’ and ‘nicotinic’ depending upon whether they were mimicked
by the then known alkaloids muscarine or nicotine. Accordingly, they were said
to be mediated by two types of cholinergic receptors, viz. muscarinic or nicotinic (N); a concept strengthened by the
finding that muscarinic actions were blocked by atropine, while nicotinic
actions were blocked by curare. In a landmark study, Ahlquist (1948) divided
adrenergic receptors into ‘α’ and ‘β’ on the basis of two
distinct rank order of potencies of adrenergic agonists. These receptors have
now been further subdivided (M1, M2 ….M5), (NM,
NN) (α1, α2) (β1, β2, β3). Multiple subtypes
of receptors for practically all transmitters, autacoids, hormones, etc. are
now known and have paved the way for introduction of numerous clinically
superior drugs. In many cases, receptor classification has provided sound
explanation for differences observed in the actions of closely related drugs.
The following criteria have been utilized in classifying
receptors:
a. Pharmacological Criteria
Classification is based on relative potencies of
selective agonists and antagonists. This is the classical and oldest approach
with direct clinical bearing; was used in delineating M and N cholinergic, α and β adrenergic, H1
and H2 histaminergic receptors, etc.
b. Tissue Distribution
The relative organ/tissue distribution is the
basis for designating the subtype, e.g. the cardiac β adrenergic receptors
as β1, while bronchial as β2. This division was
confirmed by selective agonists and antagonists as well as by molecular
cloning.
c. Ligand Binding
Measurement of specific binding of high affinity radio-labelled ligand to
cellular fragments (usually membranes) in
vitro, and its displacement by various selective agonists/antagonists is
used to delineate receptor subtypes. Multiple 5HT receptors were distinguished
by this approach. Autoradiography has helped in mapping distribution of receptor
subtypes in the brain and other organs.
d. Transducer Pathway
Receptor subtypes may
be distinguished by the
mechanism through which their activation is linked to the response, e.g. M
cholinergic receptor acts through Gproteins, while N cholinergic receptor gates
influx of Na+ ions; α adrenergic receptor acts via
IP3DAG pathway and by decreasing cAMP, while β adrenergic receptor
increases cAMP; GABAA receptor is a ligand gated Cl– channel, while
GABAB receptor increases K+ conductance through a Gprotein.
e. Molecular cloning
The receptor protein is cloned and its detailed amino acid sequence as well as
three dimentional structure is worked out. Subtypes are designated on the basis
of sequence homology. This approach has in the recent years resulted in a flood
of receptor subtypes and several isoforms (which do not differ in ligand selectivity)
of each subtype. The functional significance of many of these subtypes/
isoforms is dubious. Even receptors without known ligands (orphan receptors)
have been described.
Application
of so many approaches has thrown up several detailed, confusing and often
conflicting classifications of receptors. However, a consensus receptor
classification is now decided on a continuing basis by an expert group of the
International Union of Pharmacological Sciences (IUPHAR).
f. Silent Receptors
These
are sites which bind specific drugs but no pharmacological
response is elicited. They are better called drug acceptors or sites of
loss, e.g. plasma proteins which have binding sites for many drugs. To
avoid confusion, the term receptor should be restricted to those regulatory
binding sites which are capable of generating a response.
