Review questions and answers
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Drug development
Pharmaceutical Drugs and Dosage: Drug development - Review questions and answers
Review questions
2.1 Which of
the following is true for the drug development and regulatory process?
A. A drug’s sponsor
must submit an IND application before an FIH trial of a drug
B. An IND application must
precede an NDA submission to the FDA
C. An NDA approval must
precede a corresponding ANDA submission
D. All of the above
E. None of the above
2.2 Indicate
which of the following statements is TRUE and which is FALSE.
A. The FDA can approve
new formulations without phase III clinical trials.
B. In phase III
clinical trials, only a small number of patients are enrolled.
C. New drug substances
are extracted from plants or animals or syn-thesized in laboratories.
D. The CDER is
responsible for the approval of vaccines.
E. The ANDA requires
full clinical and nonclinical testing.
F. The ANDA can be
filed for biological products.
G. The BLA is approved
by the CBER, whereas the NDA is approved by the CDER.
2.3 A. Define the
following terminologies: FDA, IND, NDA, CDER, FIH, CBER, and BLA.
B. List the different steps involved in the drug development
and approval process.
C. In which phase of drug development are healthy subjects
evaluated?
2.4 A. What
are the specific responsibilities of the CDER and the CBER?
B.
What information does the FDA require in an IND application?
C.
What are the goals of phase I, II, and III trials?
D.
Why is the postmarketing surveillance necessary?
2.5 What are the three key components of pharmaceutical
development?
A. Bioavailability: To
ensure that the drug has reproducible and clin-ically desired bioavailability
from a dosage form.
B. Stability: To ensure
that the drug product is stable at the labeled storage conditions for the
duration of its assigned shelf life.
C. Manufacturability:
To ensure that the drug product can be manu-factured reproducibly and robustly
at a commercial scale.
D. Tolerability: To
ensure that the drug is tolerated by the subjects and there are no significant
adverse effects.
E. All of the above
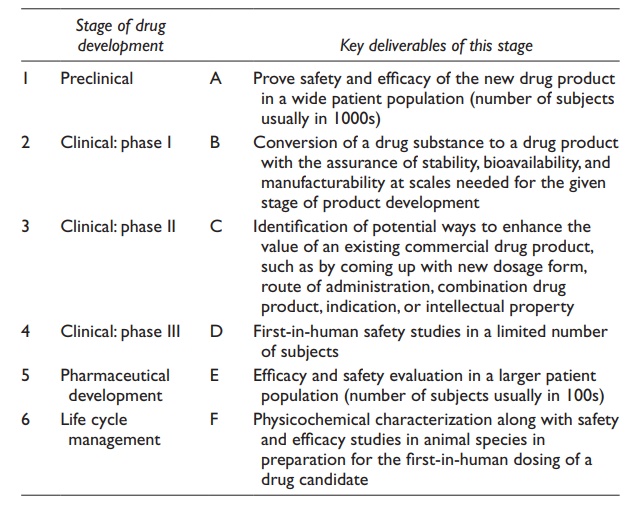
2.6 Match the stage of drug development in the left column with
the key deliverables of that stage
in the right column. Write the letter of row in the left column in front of the
corresponding row in the right column.
2.7 Interspecies dose
scaling for small-molecule compound is generally carried out using which
metric?
A. Body weight
B. Body surface area
C. Body fluid volume
D. Muscle weight
E. Fat tissue weight
2.8 Clinical studies carried out in which phase are also called
first-in-human studies?
A. Phase I
B. Phase II
C. Phase III
D. Phase IV
2.9 Which of the
following may not be a typical postcommercialization activity?
A. Investigation of a
new drug–drug combination product
B. Investigation of a
new drug–device combination product
C. First phase III
clinical trial to support commercialization of an
D. NCE
E. Investigation of an
approved drug’s ability to treat a new disease indication
Answer:
2.1 D.
2.2 A. False
B. False
C. True
D. False
E. False
F. False
G. True
2.3 A. FDA:
Food and Drug Administration; IND: investigational new drug application; NDA:
new drug application; CDER: Center for Drug Evaluation and Research; Biologics:
viruses, therapeutic serum, toxin, antitoxin, vaccines, blood, blood components
or derivatives, allergic products, or analogous products, applicable to the
preven-tion, treatment, or cure of a disease or condition of a human being.
B. Refer to the chapter.
C. Healthy subjects are evaluated in phase I clinical trials
of drug product development.
D.
A lead compound is the one that shows high bioactivity and low toxicity.
2.4 A. The CDER evaluates
prescription, generic, and OTC drug prod-ucts for safety and efficacy before
they can be marketed. It also monitors all human drugs and biopharmaceuticals
once they are in the market, and removes products from the market that may not
be manufactured properly or may cause harm to patients. The CBER regulates
biologics not reviewed by the CDER, such as vac-cines, blood and blood
products, gene therapy products, and cel-lular and tissue transplants.
B. Refer to the chapter.
C. Refer to the chapter.
D.
Postmarketing surveillance is necessary as it may contribute to the
understanding of the drug’s mechanism or scope of action, indi-cate possible
new therapeutic uses, and/or demonstrate the need for additional dosage
strengths, dosage forms, or routes of administra-tion. Post marketing
surveillance studies may also reveal additional side effects, and rare, serious
and unexpected adverse effects.
Related Topics