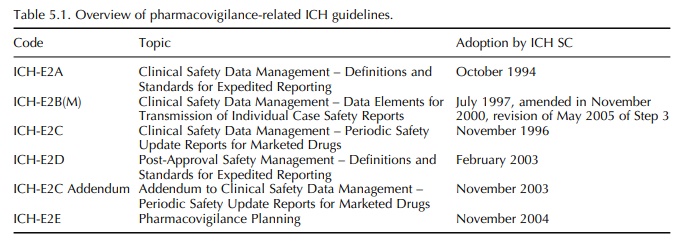
The Pharmacovigilance-Related ICH Topics
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Pharmacovigilance-Related Topics at the Level of the International Conference on Harmonisation
So far, pharmacovigilance-related topics entered the ICH process in two waves.
THE PHARMACOVIGILANCE-RELATED
ICH TOPICS
So
far, pharmacovigilance-related topics entered the ICH process in two waves. The
first wave resulted in adoption of the ICH Topic ICH-E2A in 1994 with an
extension to this work in the form of E2B and E2C, finalised between 1996 and
1997. The second wave started in 2002 with three further ICH topics, E2D, E2C
Addendum and E2E, finalised between 2003 and 2004 (Table 5.1).
Drug
Monitoring Programme established by WHO for pharmacovigilance of marketed
medicinal products.
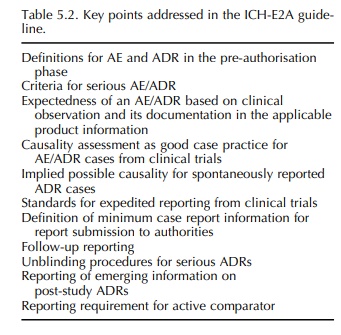
TOPIC ICH-E2A (STEP 5): CLINICAL SAFETY DATA MANAGEMENT – DEFINITIONS AND STANDARDS FOR EXPEDITED REPORTING
This
ICH guideline, adopted at ICH Step 4 in 1994, represents the first one with
relevance to pharmacovigilance. It forms part of Good Clinical Practice (GCP),
and although it deals with expedited reporting of cases of serious, unexpected
adverse drug reactions (ADRs) occurring in clinical trials during the
pre-authorisation phase, it has also been used in the post-authorisation
environment (Table 5.2). Reasons for this may have been the absence of an ICH
guideline for the post-authorisation phase, but more importantly the fact that
the ICH-E2A guideline was based on the Council for International Organi-sations
of Medical Sciences (CIOMS) I and CIOMS reports for marketed medicinal products
(CIOMS, 1990, 1992).5 The guideline also incorporated defini-tions
agreed within the framework of the International.
ICH-E2D TOPIC (STEP 5): POST-APPROVAL SAFETY MANAGEMENT – DEFINITIONS AND STANDARDS FOR EXPEDITED REPORTING
During
the second wave of pharmacovigilance-related ICH topics, it was considered
important to issue an ICH (2003a) guideline on ADR case reports specif-ically
for the post-authorisation phase (Table 5.3). Therefore, the ICH-E2D guideline
was finalised in 2003 at ICH Step 4, formalising the application of relevant
elements of ICH-E2A in the post-authorisation phase and responding to further
harmon-isation needs with regard to the definitions and management of case
reports for expedited reporting in this phase. Such further harmonisation needs
had previously been discussed in the CIOMS V Report (CIOMS, 2001), which
therefore formed an important basis for ICH-E2D.

ICH-E2B(M) TOPIC (STEP 5): CLINICAL SAFETY DATA MANAGEMENT – DATA ELEMENTS FOR TRANSMISSION OF INDIVIDUAL CASE SAFETY REPORTS
More
specifically to reporting cases of ADRs/adverse events (AEs), the ICH-E2B
guideline (ICH, 1997b) was developed to define the data fields for electronic
reporting between all stakeholders and adopted at Step 4 in 1997. Also this ICH
guideline took into account the CIOMS I Report (CIOMS, 1990). In parallel, the
M2 EWG developed the related ICH-M2 recommendations ICH-ICSR DTD (syn.: ICH-M2
E2B(M)), first also adopted at Step 4 in 1997, describing the document type
definition (DTD) of the electronic transmission of individual case safety
reports (ICSR, syn.: ADR case report). With the mandate to further improve the
definitions and spec-ifications provided in both these documents, a
Main-tenance EWG was established in 1999 and revised documents were adopted at
Step 4 in 2000 (ICH, 2000, 2001) (Table 5.4). A related questions and answers
document is being kept updated by the EWG, last revised and adopted at Step 4
in March 2005 (ICH, 2005b). To incorporate adjustments on the basis gained
through the implementation in the ICH regions, a second revision process was
initi-ated and the revised ICH-E2B(M), now called ICH-E2B(R3), was signed off
at Step 2 in May 2005 (ICH, 2005a).

ICH-E2C TOPIC (STEP 5): CLINICAL SAFETY DATA MANAGEMENT – PERIODIC SAFETY UPDATE REPORTS FOR MARKETED DRUGS
Besides
the reporting of ADR case reports in the so-called ‘expedited manner’, periodic
reporting of ADRs and other safety information was also covered in the first
wave of pharmacovigilance-related activi-ties at ICH level by adopting the
ICH-E2C guideline at Step 4 in 1996. This guideline describes the
spec-ifications for format and content of periodic safety update reports
(PSURs) reflecting the safety profile based on worldwide data and concluding
upon need for action (Table 5.5). Also ICH-E2C was based on the work achieved
by CIOMS, i.e. the CIOMS II and CIOMS III reports (CIOMS, 1992, 1995).

ICH-E2C ADDENDUM TOPIC (STEP 5): ADDENDUM TO CLINICAL SAFETY DATA MANAGEMENT – PERIODIC SAFETY UPDATE REPORTS FOR MARKETED DRUGS
After
1996, good experience had been gained with the concept of the PSURs, in
particular in the EU, and so it was agreed to promote the concept by providing
clarification and flexibility for the application of ICH-E2C in different
product types and different circum-stances (Table 5.6) (ICH, 2003b). The need
for such clarification and flexibility had been discussed before in the CIOMS V
Report (CIOMS, 2001), which was therefore used when drafting ICH-E2C Addendum.
This guideline was adopted at Step 4 in 2003.

ICH-E2E TOPIC (STEP 5): PHARMACOVIGILANCE PLANNING
This
guideline was the last one being developed during the second wave and was
adopted at Step 4 in 2004. This ICH topic was inspired by the excel-lence model
for pharmacovigilance developed in the United Kingdom with international
colleagues’ input (Waller and Evans, 2003). Also, the Japanese concept of Early
Post-Marketing Phase Vigilance (EPPV), published by the Japanese Health
Ministry in 2000 as a programme of communication between market-ing
authorisation holders and healthcare professionals on newly marketed medicinal
products to ensure safe roll-out to the market and to strengthen the
sponta-neous reporting system in the early phase of market-ing (MHW, 2000), was
considered in this context. However, pharmacovigilance planning is a different
concept; it is intended to aid marketing authorisa-tion holders and authorities
in planning data collec-tion, especially, but not exclusively, during the early
phase of marketing. Such planning is based on the so-called ‘safety
specification’, summarising identi-fied, potential and unknown risks for the
medicinal product. Various methods for data collection may be used, and ICH-E2E
therefore provides, in addition to a format for pharmacovigilance plans,
harmonised terminology for methods of active and passive surveillance as well
as principles for the conduct of pharmacoepidemiological studies of
non-experimental design (syn.: observational studies, non-interventional
studies) (Table 5.7). ICH-E2E is a framework for the formal preparation of
pharmacovig-ilance in the pre-authorisation assessment phase as well as for a
continued proactive approach through-out the post-authorisation phase. Although
ICH-E2E is not a summary of risk minimisation tools to be implemented for a
particular product, the contents of a pharmacovigilance plan may refer to such
tools, as the safety specification may depend on the risk minimisa-tion systems
in place, in particular where prescribing, dispensing and other health services
come into play. Likewise, the planned data collection methods will depend on
the health service systems and linked risk minimisation tools.

Related Topics
