Adverse Drug Reaction Databases
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Spontaneous Reporting in Germany
The two agencies in Germany have established new ADR databases in their institutions.
ADVERSE DRUG REACTION DATABASES
THE NEW SYSTEM
The
two agencies in Germany have established new ADR databases in their institutions.
As both ADR databases are essentially similar, BfArM’s database will now be
explained in more detail. After a 30-month period of development, the system
went into production in March 2005, enabling the agency to fulfil legal
reporting obligations towards the European Medicines Agency (EMEA) from May
2005 on. The system is fully compatible with international standards defined in
the International Conference on Harmoni-sation (ICH)-E2B/M2 guidelines and
supports manual data entry as well as electronic reporting according to these
standards. Controlled vocabulary and classifi-cation systems have been
implemented in accordance with EU requirements [e.g. Medical Dictionary for
Regulatory Activities (MedDRA) in its latest version for coding medical
information, ISO catalogue of country codes, WHO-Drug Dictionary and
Anatomi-cal Therapeutic Chemical (ATC) classification to deal with the huge
amount of drugs existing globally]. BfArM is aiming to have all ADR information
about individual cases covered in the new database that is seen as a major step
forwards compared with the situation so far.
The
new database is not only a data entry and storage system. In addition, a
workflow system has been implemented so that the case reports once entered into
the database, manually or electronically, can be processed through electronic
tools. This includes entry screens for single case assessment, views on the
data fields in a structured way as well as access to scanned images of
paper-based reports. In addition, standard forms for routine correspondence,
e.g. the confirmation of receipt as well as information to third parties where
appropriate, with data dynamically loaded into these forms from the database,
are available.
Furthermore,
a user interface for data retrieval exists that allows user-friendly
stratification of data. Stan-dardised entry screens to formulate routine
requests are available, but users are also allowed to perform queries on the
database without using the inter-face. Retrieval results may be presented in a
vari-ety of output reports. This includes various listings, summary tabulations
as well as graphical presenta-tions. A set of standard reports may be amended
by user-defined reports created by using a report genera-tor that is available
for those who work regularly with the database and have knowledge about the
details in more depth.
ELECTRONIC SUBMISSION
According
to EU legislation, MAHs are obliged to send reports electronically to the
responsible author-ities and the EMEA. Germany has implemented the rules in a
national regulation on the basis of the German Medicines Act. This national
regulation became valid on 20 October 2005, and it offers the possibility of
switching to electronic reporting not at a defined date but over a period
giving companies as well as regulators the chance to cope with challenges the
new technology imposes.
Companies
with a very low number of reports per year, defined as less than an average of
ten reports annually during a period of the last 5 years, may apply for a
waiver that allows paper-based reporting despite the legal obligation for
electronic transmission. In this case, most reports sent to BfArM or PEI from
those companies are closely monitored by both institutes. The waiver may be
withdrawn if the number of reports exceed the limits in the future.
Companies
that are obliged to report electronically have to undergo a test phase in which
cases are submitted to a database that is only designed for user tests and
developmental purposes. The tests are focused on technical aspects as well as
on content of and coding in the electronic reports. These data are compared
with the information provided in the paper forms normally sent. After
successful comple-tion of this test that is structured similarly to the EMEA
test scenario, companies shall enter into the so-called ‘production phase’.
BfArM has started this phase of transmitting case reports only electronically
in December 2005 with three companies and has now registered about 50 companies
for electronic report-ing. The first phase of the transition period towards
electronic reporting is focused on the major companies with a high number of
reports so that BfArM is able to handle the huge amount of data better than
before. The proportion of reports submitted by these large companies = 70 is expected to equal about 95%
of all reports per year. PEI has been receiving reports electronically since
February 2005, presently from 20 companies. On a daily basis, BfArM and PEI
forward all new reports in their databases electronically to the EudraVigilance
database run by the EMEA.
Problems
that can be seen after the first months of experience are data inconsistencies
across data fields and data coding that appear to be a challenge in the context
of the new rules. So far, companies were obliged to transmit all relevant
information in accor-dance with the legal time lines. This allows the
provi-sion of data in an unstructured way even if put into appropriate report
forms. The new obligations, laid down in ICH guidelines that are referenced by
the applicable EU documents, go beyond these require-ments stating that
information in the narrative should be reflected by accurate coding in the
appropriate data fields (ICH E2D guideline). Therefore, the obligation is not
only to provide but also to structure informa-tion according to the agreed
international standards facilitating data retrieval to find the legendary
‘needle in the haystack’. Thus, BfArM sees its role not only in dealing with
the new technologies and the huge number of reports but also in monitoring
whether the requirements of structuring data are fulfilled and in providing
feedback accordingly.
REPORT NUMBERS
With
regard to actual figures, BfArM receives about 17 000 national case reports per
year, not counting duplicate reporting and follow-ups. Report numbers from
foreign countries, EU as well as non-EU, are currently declining and amount to
about 120 000 annually, again not counting duplicate reporting and follow-ups.
Declining numbers during the past year are because of the legal implementation
of the revised EU rules for ADR reporting outlined in the so-called ‘Review
2004’. Most cases are thus received from countries outside the EU.
Unlike
in other countries, BfArM receives most domestic cases through pharmaceutical
companies. As far as national reports are concerned, 85% of the incoming
information derive from this source. The second largest number of reports is
received from drug commissions of healthcare professionals that exist for
physicians (see above) as well as for pharmacists and dentists. These sources
provide about 10% of national reports with the Drug Commission of the German
Medical Association being the most important one. The remainder of cases is
received from physicians directly (including from investigator-initiated
trials). Direct consumer reports, i.e. reports from patients or their
relatives, amount to an only very low number (about 100 per year). This low
number sounds surpris-ing because topics of drug safety are often discussed
publicly and intensively, including in lay media. On the contrary, direct
consumer reporting is not encour-aged by our institute. The experience over the
past decades has not shown that information has been lost by not having
encouraged consumer reporting, i.e. the information has been received through
other routes. The BfArM tends more to follow the interna-tional recommendations
that patients should see their doctors first to seek medical advice and to
trans-mit well-documented case reports that are the result of collaboration
between patient and physician. This strategy is encouraged by BfArM and PEI
whenever appropriate.
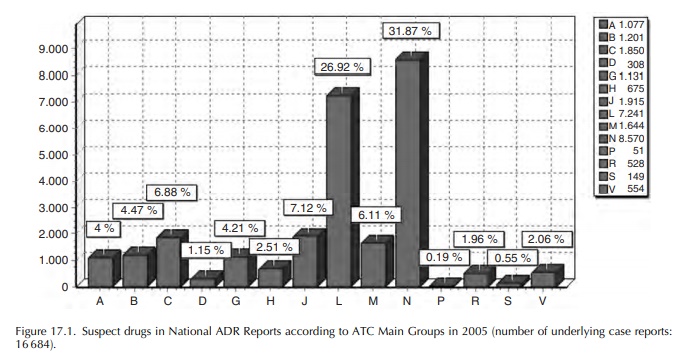
Most
of the ADR reports refer to drugs used in cancer treatment (main ATC Group L)
and those used in neurology or as analgesics (main ATC Group N). The
distribution of suspect/interacting drugs mentioned in case reports – maybe
more than one per case – according to the ATC classification is shown in the
following figure for the year 2005 (Figure 17.1).
Related Topics
