Transducer Mechanisms
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacodynamics Mechanism Of Drug Action; Receptor Pharmacology
Considerable progress has been made in the understanding of transducer mechanisms which in most instances have been found to be highly complex multistep processes that provide for amplification and integration of concurrently received extra and intracellular signals at each step.
TRANSDUCER MECHANISMS
Considerable progress
has been made in the understanding of transducer mechanisms which in most
instances have been found to be highly complex multistep processes that provide
for amplification and integration of concurrently received extra and intracellular
signals at each step. Because only a handful of transducer pathways are shared
by a large number of receptors, the cell is able to generate an integrated
response reflecting the sum total of diverse signal input. The transducer
mechanisms can be grouped into 4 major categories. Receptors falling in one category
have also been found to possess considerable structural homology, and belong to
one super family of receptors.
1. Gprotein Coupled Receptors (GPCR)
These are a large
family of cell membrane receptors which are linked to the effector (enzyme/
channel/carrier protein) through one or more GTPactivated proteins (Gproteins)
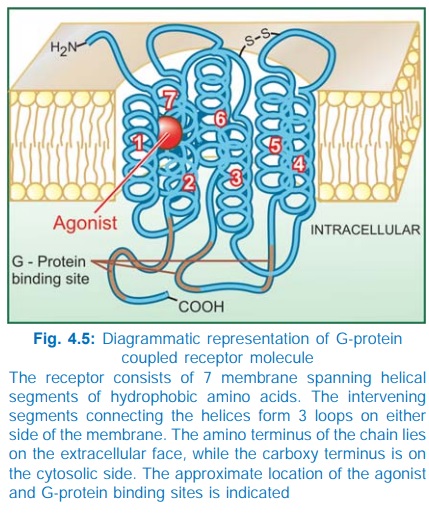
for response effectuation. All such receptors have a common pattern of
structural organization (Fig. 4.5). The molecule has 7 αhelical membrane
spanning hydrophobic amino acid (AA) segments which run into 3 extracellular
and 3 intracellular loops. The agonist binding site is located somewhere
between the helices on the extracellular face, while another recognition site
formed by cytosolic segments binds the coupling Gprotein. The Gproteins float
in the membrane with their exposed domain lying in the cytosol, and are
heterotrimeric in composition (α, β and γ subunits). In the inactive state GDP is bound
to their exposed domain; activation through the receptor leads to displacement
of GDP by GTP. The active αsubunit carrying GTP dissociates from the
other two subunits and either activates or inhibits the effector. The βγ subunits have also
been shown to modulate certain effectors like receptoroperated K+ channels,
adenylylcyclase (AC) and phospholipase C.
A
number of G proteins distinguished by their α subunits have been
described. The important ones with their action on the effector are:
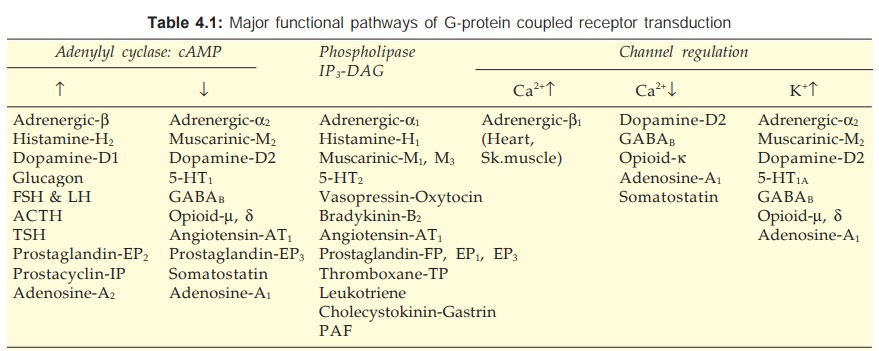
Gs : Adenylyl cyclase ↑, Ca2+ channel ↑
Gi : Adenylyl cyclase ↓, K+ channel ↑
Go : Ca2+ channel ↓
Gq : Phospholipase C ↑
G13 : Na+/H+ exchange ↑
In
addition Gn, Gk, Gt and Golf have been distinguished. A limited
number of Gproteins are shared between different receptors and one receptor can
utilize more than one Gprotein (agonist pleotropy), e.g. the following couplers
have been associated with different receptors.
Receptor Coupler
Muscarinic Gi, Go, Gq
Dopamine D2 Gi, Go
βadrenergic Gs, Gi
α1adrenergic Gq
α2adrenergic Gi, Gs, Go
GABAB Gi, Go
5HT Gi, Gq, Gs, Gk
In addition, a
receptor can utilize different biochemical pathways in different tissues.
The α-subunit has GTPase
activity: the bound GTP is slowly hydrolysed to GDP: the αsubunit then dissociates
from the effector to rejoin its other subunits, but not before the effector has
been activated/inhibited for a few seconds and the signal has been amplified.
The onset time of response through this type of receptors is also in seconds.
There are three major
effector pathways (Table 4.1) through which GPCRs function.
a) Adenylyl cyclase: cAMP pathway Activation of AC results in intracellular accumulation of
second messenger cAMP (Fig. 4.6) which functions mainly through cAMPdependent
protein kinase (PKA). The PKA phosphorylates and alters
the function of many enzymes, ion channels, transporters and structural
proteins to manifest as increased contractility/impulse generation (heart),
relaxation (smooth muscle), glycogenolysis, lipolysis, inhibition of secretion/mediator
release, modulation of junctional transmission, hormone synthesis, etc. In
addition, cAMP directly opens a specific type of membrane Ca2+ channel called cyclic nucleotide gated channel (CNG) in
the heart, brain and kidney. Responses opposite to the above are produced when
AC is inhibited through inhibitory G-protein.
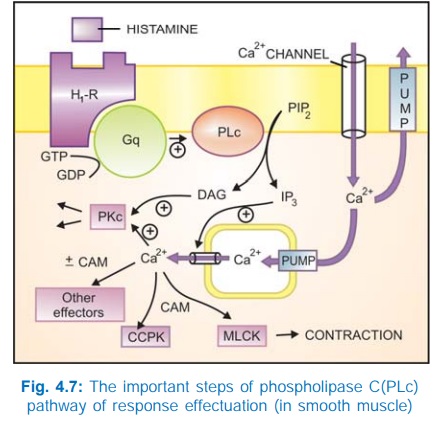
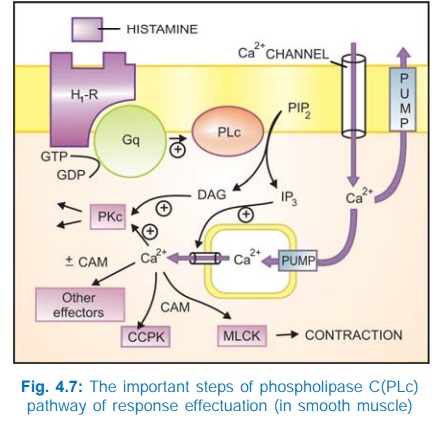
b) Phospholipase C: IP3DAG pathway Activation of phospholipase
C (PLc) hydrolyses the membrane phospholipid phosphatidyl inositol 4, 5bisphosphate
(PIP2) to generate the second messengers inositol 1,4,5trisphosphate
(IP3) and diacylglycerol (DAG). The IP3 mobilises Ca2+
from intracellular organellar depots and DAG enhances protein kinase C (PKc)
activation by Ca2+ (Fig. 4.7). Cytosolic Ca2+ (third messenger in this setting)
is a highly versatile regulator acting through calmodulin (CAM), PKc and other
effectors—mediates/modulates contraction, secretion/transmitter release,
eicosanoid synthesis, neuronal excitability, intracellular movements, membrane
function, metabolism, cell proliferation, etc. Like AC, the PLc can also be
inhibited through inhibitory Gprotein when directionally opposite responses would
be expected.
Intracellular Ca2+
release has been found to occur in waves (Ca2+ mediated Ca2+ release from
successive pools facilitated by inositol 1, 3, 4, 5tetrakisphosphate—IP4)
and exhibits a variety of agonist and concentration dependent oscillatory
patterns. The activation of different effectors may depend on the amplitude and
pattern of these oscillations. Thus, the same intracellular messenger can
trigger different responses depending on the nature and strength of the
extracellular signal.
c) Channel regulation The activated Gproteins
can also open or close ionic channels specific for Ca2+, K+ or Na+, without the
intervention of any second messenger like cAMP or IP3, and bring
about hyperpolarization/depolarization/ changes in intracellular Ca2+. The Gs
opens Ca2+ channels in myocardium and skeletal muscles, while Gi and Go open K+
channels in heart and smooth muscle as well as close neuronal Ca2+ channels.
Physiological responses like changes in inotropy, chronotropy, transmitter
release, neuronal activity and smooth muscle relaxation follow. Receptors found
to regulate ionic channels through Gproteins are listed in Table 4.1.
2. Receptors With Intrinsic Ion
Channel
These cell surface receptors, also called ligand gated ion channels, enclose
ion selective channels (for Na+, K+,
Ca2+ or Cl¯) within their molecules. Agonist binding opens the channel (Fig.
4.4) and causes depolarization/hyperpolarization/ changes in cytosolic ionic
composition, depending on the ion that flows through. The nicotinic
cholinergic, GABAA, glycine (inhibitory), excitatory AA (kainate,
NMDA or NmethylDaspartate, quisqualate) and 5HT3 receptors fall in
this category.
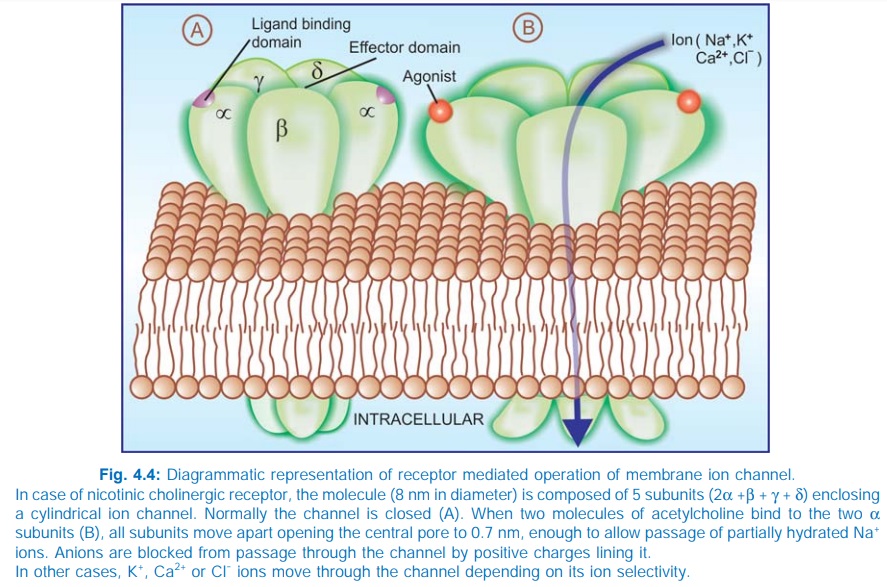
The receptor is usually a pentameric protein; all subunits, in
addition to large intra and extracellular segments, generally have four membrane
spanning domains in each of which the AA chain traverses the width of the
membrane six times. The subunits are thought to be arranged round the channel
like a rosette and the α subunits usually bear the agonist binding
sites.
Certain receptoroperated (or ligandgated) ion
channels also have secondary ligands which bind to an allosteric site and
modulate the gating of the channel by the primary ligand, e.g. the benzodiazepine
receptor modulates GABAA gated Cl¯channel.
Thus, in these receptors, agonists directly
operate ion channels, without the intervention of any coupling protein or
second messenger. The onset and offset of responses through this class of
receptors is the fastest (in milliseconds).
3. Enzyme-Linked Receptors
This class of receptors have a subunit with
enzymatic property or bind a JAK (Janus Kinase) enzyme on activation. The
agonist binding site and the catalytic site lie respectively on the outer and
inner face of the plasma membrane (Fig. 4.8). These two domains are
interconnected through a single transmembrane stretch of peptide chain. There
are two major subgroups of such receptors.
a. Those that have intrinsic enzymatic
activity.
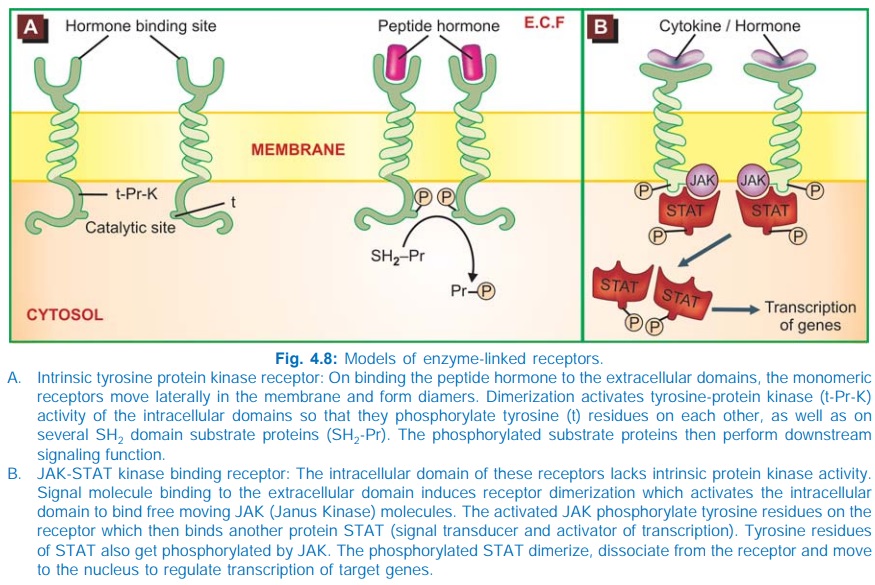
b. Those that lack intrinsic enzymatic
activity, but bind a JAKSTAT kinase on activation.
a. Intrinsic Enzyme Receptors
The intracellular
domain is either a protein kinase or guanylyl cyclase.
In
most cases the protein kinase specifically phosphorylates tyrosine residues on
substrate proteins (Fig. 4.8A), e.g. insulin, epidermal growth factor (EGF),
nerve growth factor (NGF) receptors, but in few it is a serine or threonine
protein kinase. In the monomeric state, the kinase remains inactive. Agonist
binding induces dimerization of receptor molecules and activates the kinase to
autophosphorylate tyrosine residues on each other, increasing their affinity
for binding substrate proteins and carrying forward the cascade of tyrosine
phosphorylations. Intracellular events are triggered by phosphorylation of
relevant proteins, many of which carry a SH2 domain. A general
feature of this class of receptors is that their dimerization also promotes
receptor internalization, degradation in lysosomes and down regulation.
The
enzyme can also be guanylyl cyclase (GC), as in the case of atrial natriuretic
peptide (ANP). Agonist activation of the receptor generates cGMP in the cytosol
as a second messenger which in turn activates cGMPdependent protein kinase and
modulates cellular activity.
b. JAK-STAT Kinase Binding Receptors
These receptors differ in
not having any intrinsic catalytic domain. Agonist induced dimerization alters
the intracellular domain conformation to increase its affinity for a cytosolic
tyrosine protein kinase JAK. On binding, JAK gets activated and phosphorylates
tyrosine residues of the receptor, which now binds another free moving protein
STAT (signal transducer and activator of transcription) which is also phosphorylated
by JAK. Pairs of phosphorylated STAT dimerize and translocate to the nucleus to
regulate gene transcription resulting in a biological response. Many cytokines,
growth hormone, interferons, etc. act through this type of receptor.
The
enzymelinked receptors transduce responses in a matter of few minutes to a few
hours.
4. Receptors
Regulating Gene Expression (Transcription Factors)
In
contrast to the above 3 classes of receptors, these are intracellular
(cytoplasmic or nuclear) soluble proteins which respond to lipid soluble chemical
messengers that penetrate the cell (Fig. 4.9). The receptor protein (specific
for each hormone/ regulator) is inherently capable of binding to specific
genes, but is kept inhibited till the hormone binds near its carboxy terminus
and exposes the DNA binding regulatory segment located in the middle of the
molecule. Attachment of the receptor protein to the genes facilitates their
expression so that specific mRNA is synthesized on the template of the gene.
This mRNA moves to the ribosomes and directs synthesis of specific proteins
which regulate the activity of target cells.
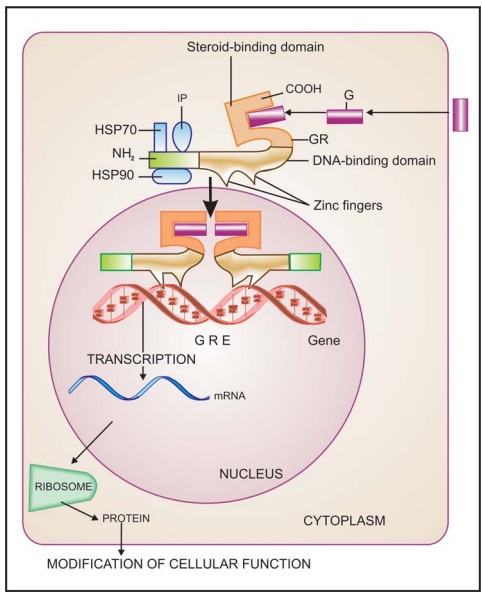

All steroidal hormones
(glucocorticoids, mineralocorticoids, androgens, estrogens, progesterone),
thyroxine, vit D and vit A function in this manner. Different steroidal
hormones affect different target cells and produce different effects because
each one binds to its own receptor and directs a unique pattern of synthesis of
specific proteins. The specificity as to which hormone will be bound is
provided by the hormone binding domain, while that as to which gene will be
activated or repressed is a function of the DNA binding/Nterminus domain.
Chimeric receptors have been produced which respond to one hormone, but produce
the effects of the other hormone.
This transduction
mechanism is the slowest in its time course of action (takes hours).
Receptor Regulation
Receptors exist in a
dynamic state; their density and efficacy is subject to regulation by the level
of on going activity, feedback from their own signal output and other physio-pathological
influences. In tonically active systems, prolonged deprivation of the agonist
(by denervation or continued use of an antagonist or a drug which reduces
input) results in super sensitivity of the receptor as well as the effector
system to the agonist. This has clinical relevance in clonidine/CNS depressant/
opioid withdrawal syndromes, sudden discontinuation of propranolol in angina
pectoris, etc. The mechanisms involved may be unmasking of receptors or their
proliferation (up regulation) or
accentuation of signal amplification by the transducer.
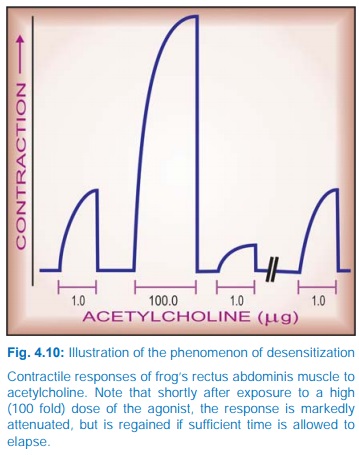
Conversely,
continued/intense receptor stimulation causes desensitization or
refractoriness: the receptor becomes less sensitive to the agonist. This can be
easily demonstrated experimentally (Fig. 4.10); clinical examples are bronchial
asthma patients treated continuously with β adrenergic agonists
and parkinsonian patients treated with high doses of levodopa. The changes may
be brought about by:
i) Masking or internalization of the receptor (it becomes
inaccessible to the agonist). In this case refractoriness develops as well as
fades quickly.
In the case of β adrenergic receptor,
it has been found that agonist binding promotes phosphorylation of its serine
residues near the intracellular carboxy terminus by an enzyme β adrenergic receptor
kinase (βARK), allowing it to
bind a protein called βarrestin which hinders its interaction with Gs
→ receptor transduction
is impaired. When the βagonist is removed, the serine residues are
dephosphorylated and receptor mediated activation of Gs is restored.
ii) Decreased synthesis/increased destruction of the receptor (down regulation): refractoriness develops
over weeks or months and recedes slowly. Receptor down regulation is
particularly exhibited by the tyrosine protein kinase receptors.
Some times response to
all agonists which act through different receptors but produce the same overt
effect (e.g. histamine and acetylcholine both contract intestinal smooth
muscle) is decreased by exposure to any one of these agonists (heterologous desensitization),
showing that mechanisms of response effectuation have become less efficient.
However, often desensitization is limited to agonists of the receptor being
repeatedly activated (homologous desensitization).
Both
homologous and heterologous desensitization has been observed in the case of
GPCRs. The BARKβ arrestin mechanism described above produces homologous
desensitization. The GPCRs transduce many responses by activating PKA
and PKC. These kinases phosphorylate the GPCRs as well rather non-selectively
(at a site different from that of BARK) and hinder their interaction with G-proteins,
resulting in heterologous desensitization.
Related Topics
