Kinetics of Elimination
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacokinetics; Metabolism Excretion Of Drugs, Kinetics Of Elimination
The knowledge of kinetics of elimination of a drug provides the basis for, as well as serves to devise rational dosage regimens and to modify them according to individual needs. There are three fundamental pharmacokinetic parameters, viz. bioavailability (F), volume of distribution (V) and clearance (CL) which must be understood. The first two have already been considered.
KINETICS OF ELIMINATION
The knowledge of
kinetics of elimination of a drug provides the basis for, as well as serves to
devise rational dosage regimens and to modify them according to individual
needs. There are three fundamental pharmacokinetic parameters, viz.
bioavailability (F), volume of
distribution (V) and clearance (CL) which must be understood. The first
two have already been considered.
Drug
elimination is the sumtotal of metabolic inactivation and excretion. As
depicted in Fig. 2.1, drug is eliminated only from the central compartment
(blood) which is in equilibrium with peripheral compartments including the site
of action. Depending upon the ability of the body to eliminate a drug, a
certain fraction of the central compartment may be considered to be totally
‘cleared’ of that drug in a given period of time to account for elimination
over that period.
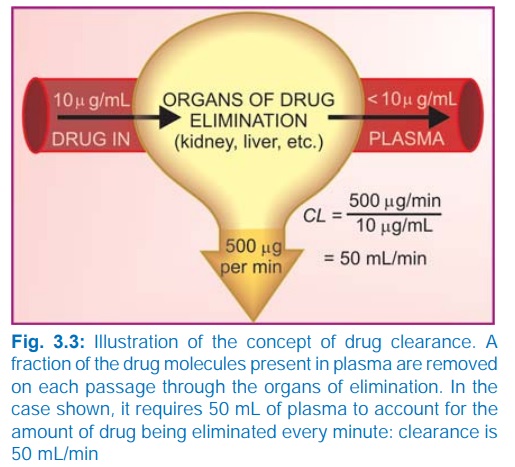
Clearance (CL)
The clearance of a
drug is the theoretical volume of
plasma from which the drug is completely removed in unit time (analogy creatinine
clearance; Fig. 3.3). It can be calculated as
CL = Rate of elimination/C ...(1)
where C is the plasma concentration.
For majority of drugs
the processes involved in elimination are not saturated over the clinically
obtained concentrations, they follow:
First order (exponential) kinetics
The rate of elimination is directly proportional to the drug
concentration, CL remains constant;
or a constant fraction of the drug
present in the body is eliminated in
unit time.
Few drugs, however,
saturate eliminating mechanisms and are handled by—
Zero order (linear) kinetics The rate of elimination
remains constant irrespective of drug concentration, CL decreases with increase in concentration; or a constant amount of the drug is eliminated in unit
time, e.g. ethyl alcohol.
The elimination of
some drugs approaches saturation over the therapeutic range, kinetics changes
from first order to zero order at higher doses. As a result plasma
concentration increases disproportionately with increase in dose, (See Fig. 3.5) as occurs in case of phenytoin,
tolbutamide, theophylline, warfarin.
Plasma half life
The Plasma halflife
(t½) of a drug is the time taken
for its plasma concentration to be reduced to half of its original value.
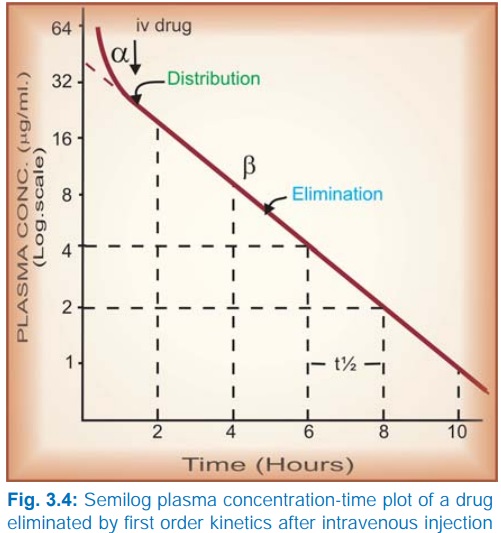
Taking
the simplest case of a drug which has rapid one compartment distribution and
first order elimination, and is given i.v. a semilog plasma concentrationtime
plot as shown in Fig. 3.4 is obtained. The plot has two slopes.
§ initial rapidly
declining (α) phase—due to
distribution.
§ later less declined (β) phase—due to elimination.
At
least two halflives (distribution t½ and elimination t½) can be calculated from
the two slopes. The elimination half life derived from the slope is simply
called the ‘half life’ of the drug.
Most
drugs infact have multicompartment distribution and multiexponential decay of
plasma concentrationtime plot. Halflives calculated from the terminal slopes
(when plasma concentrations are very low) are exceptionally long, probably due
to release of the drug from slow equilibrating tissues, enterohepatic
circulation, etc. Only the t½ calculated over the therapeutic plasma
concentration range is clinically relevant. It is this t½ which is commonly
mentioned.
Mathematically, elimination
t½ is
ln2
t½ = —— ...(2)
k
Where ln2 is the
natural logarithm of 2 (or 0.693) and k
is the elimination rate constant of
the drug, i.e. the fraction of the total amount of drug in the body which is
removed per unit time. For example, if 2 g of the drug is present in the body
and 0.1 g is eliminated every hour, then k
= 0.1/2 = 0.05 or 5% per hour. It is calculated as:
CL
k = —– ...(3)
V
V
therefore t½ = 0.693 × —— ...(4)
C L
As such, halflife is a
derived parameter from two variables V
and CL both of which may change
independently. It, therefore, is not an exact index of drug elimination.
Nevertheless, it is a simple and useful guide to the sojourn of the drug in the
body, i.e. after
1
t½ – 50% drug is eliminated.
2
t½ – 75% (50 + 25) drug is eliminated.
3
t½ – 87.5% (50 + 25 + 12.5) drug is eliminated.
4 t½ – 93.75% (50 + 25 + 12.5 + 6.25) drug is eliminated.
Thus, nearly complete drug elimination occurs
in 4–5 half lives.
For drugs eliminated by—
First order kinetics—t½ remains constant because
V and CL do not change with dose.
Zero order kinetics—t½ increases with dose
because CL progressively decreases as
dose is increased.
Repeated drug administration
When a drug is repeated at relatively short
intervals, it accumulates in the body until elimination balances input and a steady state plasma concentration (Cpss) is attained—

From this equation it is implied that doubling
the dose rate would double the average Cpss
and so on. Further, if the therapeutic plasma concentration of the drug has
been worked out and its CL is known,
the dose rate needed to achieve the target
Cpss can be determined—
dose rate = target Cpss ×
CL
...(6)
After oral administration, often only a
fraction (F) of the dose reaches
systemic circulation in the active form. In such a case—
target Cpss × CL
dose rate = ———————— ...(7)
F
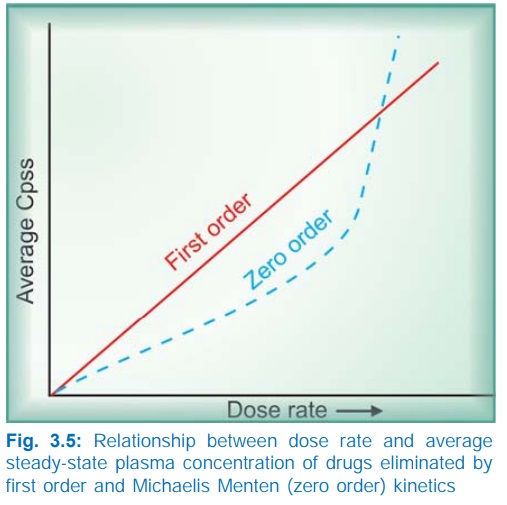
The dose rate Cpss relationship is linear only in case
of drugs eliminated by first order kinetics. For drugs (e.g. phenytoin) which
follow Michaelis Menten kinetics, elimination changes from first order to zero
order kinetics over the therapeutic range. Increase in their dose beyond
saturation levels causes an increase in Cpss
which is out of proportion to the change in dose rate (Fig. 3.5). In their
case:
(Vmax)
(C)
Rate of drug
elimination = ————— ...(8)
Km + C
where C is the plasma concentration of the
drug, Vmax is the maximum
rate of drug elimination, and Km
is the plasma concentration at which elimination
rate is half maximal.
Plateau Principle
When
constant dose of a drug is repeated before the expiry of 4 t½, it would achieve
higher peak concentration, because some remnant of the previous dose will be
present in the body. This continues with every dose until progressively
increasing rate of elimination (which increases with increase in concentration)
balances the amount administered over the dose interval. Subsequently plasma
concentration plateaus and fluctuates about an average steadystate level. This
is known as the plateau principle of drug accumulation. Steadystate is reached
in 4–5 half lives unless dose interval is very much longer than t½ (Fig. 3.6).
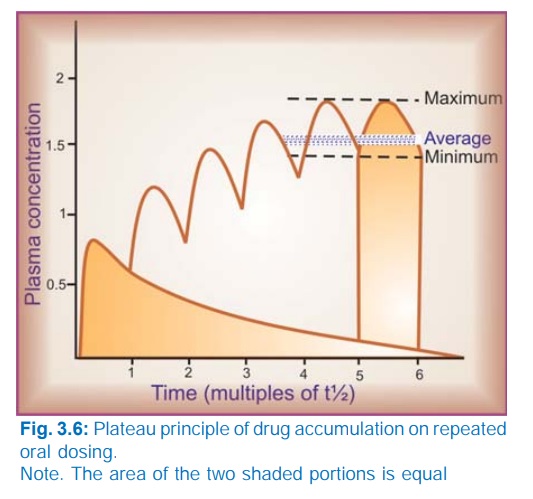
The amplitude of fluctuations in plasma concentration at steadystate depends on the dose interval relative to the t½, i.e. the difference between the maximum and minimum levels is less if smaller doses are repeated more frequently (dose rate remaining constant). Dose intervals are generally a compromise between what amplitude of fluctuations is clinically tolerated (loss of efficacy at troughs and side effects at peaks) and what frequency of dosing is convenient. However, if the dose rate is changed, a new average Cpss is attained over the next 4–5 half lives. When the drug is administered orally (absorption takes some time), average Cpss is approximately 1/3 of the way between the minimal and maximal levels in a dose interval.
Target Level Strategy
For drugs whose
effects are not easily
quantifiable and safety margin is not big, e.g. anticonvulsants,
antidepressants, lithium, antiarrhythmics, theophylline, some antimicrobials,
etc. or those given to prevent an event, it is best to aim at achieving a
certain plasma concentration which has been defined to be in the therapeutic
range; such data are now available for most drugs of this type.
Drugs
with short t½ (upto 2–3 hr) administered at conventional intervals (6–12 hr)
achieve the target levels only intermittently and fluctuations in plasma
concentration are marked. In case of many drugs (penicillin, ampicillin,
chloramphenicol, erythromycin, propranolol) this however is therapeutically
acceptable.
For
drugs with longer t½ a dose that is sufficient to attain the target
concentration after single administration, if repeated will accumulate according
to plateau principle and produce toxicity later on. On the other hand, if the
dosing is such as to attain target level at steady state, the therapeutic
effect will be delayed by about 4 half lives (this may be clinically
unacceptable). Such drugs are often administered by initial loading and
subsequent maintenance doses.
Loading Dose
This is a single or
few quickly repeated doses given
in the beginning to attain target concentration rapidly. It may be calculated
as—
target
Cp × V
Loading dose = —————— ...(9)
F
Thus, loading dose is
governed only by V and not by CL or t½.
Maintenance Dose
This dose is one that
is to be repeated at
specified intervals after the attainment of target Cpss so as to maintain the same by balancing elimination. The
maintenance dose rate is computed by equation (7) and is governed by CL (or t½) of the drug. If facilities
for measurement of drug concentration are available, attainment of target level
in a patient can be verified subsequently and dose rate adjusted if required.
Such two phase dosing
provides rapid therapeutic effect with long term safety; frequently applied to
digoxin, chloroquine, long acting sulfonamides, doxycycline, amiodarone, etc.
However, if there is no urgency, maintenance doses can be given from the
beginning. The concept of loading and maintenance dose is valid also for short
t½ drugs and i.v. administration in critically ill patients, e.g. lidocaine (t½
1.5 hr) used for cardiac arrhythmias is given as an i.v. bolus dose followed by
slow i.v. infusion or intermittent fractional dosing.
Monitoring Of Plasma Concentration Of Drugs
It is clear from the
above considerations that the Cpss of
a drug attained in a given patient depends
on its F, V and CL in that patient. Because each of
these parameters varies considerably among individuals, the actual Cpss in a patient may be 1/3 to 3 times
that calculated on the basis of population data. Measurement of plasma drug
concentration can give an estimate of the pharmacokinetic variables in that
patient and the magnitude of deviation from the ‘average patient’, so that
appropriate adjustments in the dosage regimen can be made.
In case of drugs
obeying first order kinetics:

Therapeutic
drug monitoring (TDM) is particularly useful in the following situations:
Drugs with low safety margin—digoxin, anticonvulsants,
antiarrhythmics, theophylline, aminoglycoside antibiotics, lithium, tricyclic
antidepressants.
If individual variations are large—antidepressants, lithium.
Potentially toxic drugs used in the presence of renal
failure—aminoglycoside antibiotics, vancomycin.
In case of poisoning.
In case of failure of response without any apparent
reason—antimicrobials.
To check patient compliance—psychopharmacological agents.
Selection
of the correct interval between drug administration and drawing of blood sample
for TDM is critical, and depends on the purpose of TDM as well as the nature of
the drug.
a. When
the purpose is dose adjustment: In case of drugs which need to act continuously
(relatively longacting drugs), it is prudent to measure the trough steadystate
blood levels, i.e. just prior to the next dose, because this is governed by
both V and CL. On the other hand, for shortacting drugs which achieve
therapeutic levels only intermittently (e.g. ampicillin, gentamicin), sampling
is done in the immediate postabsorptive phase (usually after 1–2 hours of oral/
i.m. dosing) to reflect the peak levels.
In case of poisoning: Blood for drug level
estimation should be taken at the earliest and then repeatedly to confirm the
poisoning and to monitor the progress.
For checking compliance to medication: Even
random blood sampling can be informative.
Monitoring Of Plasma Concentration Is Of No Value For
§ Drugs whose response
is easily measurable— antihypertensives, hypoglycaemics, diuretics, oral
anticoagulants, general anaesthetics.
§ Drugs activated in the
body—levodopa.
§ ‘Hit and run drugs’
(whose effect lasts much longer than the drug itself)—reserpine, guanethidine,
MAO inhibitors, omeprazole.
§ Drugs with
irreversible action—organophosphate anticholinesterases, phenoxybenzamine.
