Aminoglycoside Antibiotics
| Home | | Pharmacology |Chapter: Essential pharmacology : Aminoglycoside Antibiotics
These are a group of natural and semisynthetic antibiotics having polybasic amino groups linked glycosidically to two or more aminosugar (streptidine, 2deoxy streptamine, garosamine) residues.
AMINOGLYCOSIDE ANTIBIOTICS
These are a group of
natural and semisynthetic antibiotics having polybasic amino groups linked
glycosidically to two or more aminosugar (streptidine, 2deoxy streptamine,
garosamine) residues.
Unlike penicillin,
which was a chance discovery, aminoglycosides are products of deliberate search
for drugs effective against gram-negative bacteria. Streptomycin was the first member discovered in 1944 by Waksman and
his colleagues. It assumed great importance because it was active against
tubercle bacilli. Others have been produced later; now aminoglycosides are a
sizable family. All aminoglycosides are produced by soil actinomycetes and have
many common properties.
Systemic Aminoglycosides
Streptomycin
Amikacin
Gentamicin
Sisomicin
Kanamycin
Netilmicin
Tobramycin
Topical Aminoglycosides
Neomycin
Framycetin
Common Properties Of
Aminoglycoside Antibiotics
· All are used as
sulfate salts, which are highly water soluble; solutions are stable for months.
· They ionize in solution;
are not absorbed orally; distribute only extracellularly; do not penetrate
brain or CSF.
· All are excreted
unchanged in urine by glomerular filtration.
· All are bactericidal
and more active at alkaline pH.
· They act by
interferring with bacterial protein synthesis.
· All are active
primarily against aerobic gram-negative bacilli and do not inhibit anaerobes.
· There is only partial
cross resistance among them.
· They have relatively
narrow margin of safety.
· All exhibit ototoxicity
and nephrotoxicity.
Mechanism Of Action
The aminoglycosides
are bactericidal antibiotics, all having the same general pattern of action which
may be described in two main steps:
a) Transport of the
aminoglycoside through the bacterial cell wall and cytoplasmic membrane.
b) Binding to
ribosomes resulting in inhibition of protein synthesis.
Transport of aminoglycoside
into bacteria is a multistep process. They diffuse across the outer coat of gram-negative
bacteria through porin channels. Entry from the periplasmic space across the
cytoplasmic membrane is carrier mediated which is linked to the electron transport
chain. Thus, penetration is dependent upon maintenance of a polarized membrane
and on oxygen dependent active processes. These processes are inactivated under
anaerobic conditions; anaerobes are not sensitive and facultative anaerobes are
more resistant when O2 supply is deficient, e.g. inside big
abscesses. Penetration is also favoured by high pH; aminoglycosides are ~20
times more active in alkaline than in acidic medium. Inhibitors of bacterial
cell wall (βlactams, vancomycin)
enhance entry of aminoglycosides and exhibit synergism.
Once inside the bacterial cell, streptomycin binds to 30S
ribosomes, but other aminoglycosides bind to additional sites on 50S subunit,
as well as to 30S50S interface. They freeze initiation of protein synthesis, prevent
polysome formation and promote their disaggregation to monosomes so that only
one ribosome is attached to each strand of mRNA. Binding of aminoglycoside to
30S50S juncture causes distortion of mRNA codon recognition resulting in misreading
of the code: one or more wrong amino acids are entered in the peptide chain
and/ or peptides of abnormal lengths are produced. Different aminoglycosides
cause misreading at different levels depending upon their selective affinity
for specific ribosomal proteins.
The cidal action of these drugs appears to be based on secondary
changes in the integrity of bacterial cell membrane, because other antibiotics
which inhibit protein synthesis (tetracyclines, chloramphenicol, erythromycin)
are only static. After exposure to aminoglycosides, sensitive bacteria become
more permeable; ions, amino acids and even proteins leak out followed by cell
death. This probably results from incorporation of the defective proteins into
the cell membrane. One of the consequences of aminoglycoside induced alteration
of cell membrane is augmentation of the carrier-mediated entry of the antibiotic.
This reinforces the lethal action.
The cidal action of
aminoglycosides is concentration dependent, i.e. rate of bacterial cell killing
is directly related to the ratio of the peak antibiotic concentration to the MIC
value. They also exert a long and concentration dependent ‘post-antibiotic
effect’. It has, therefore, been argued that despite their short t½ (2–4 hr),
single injection of the total daily dose of aminoglycoside may be more effective
and possibly less toxic than its conventional division into 2–3 doses.
Mechanism Of Resistance
Resistance to
aminoglycosides is acquired by one of the following mechanisms:
a)
Acquisition of cell membrane bound inactivating
enzymes which phosphorylate/ adenylate or acetylate the antibiotic. The conjugated
aminoglycosides do not bind to the target ribosomes and are incapable of
enhancing active transport like the unaltered drug. These enzymes are acquired
mainly by conjugation and transfer of plasmids. Nosocomial microbes have become
rich in such plasmids, some of which encode for multidrug resistance. This is
the most important mechanism of development of resistance to aminoglycosides.
Susceptibility of different aminoglycosides to these enzymes differs. Thus,
cross resistance among different members is partial or absent.
b)
Mutation decreasing the affinity of ribosomal
proteins that normally bind the aminoglycoside: this mechanism can confer high
degree resistance, but operates to a limited extent, e.g. E. coli that develop streptomycin resistance by single step
mutation do not bind the antibiotic on the polyribosome. Only a few other
instances are known. This type of resistance is specific for a particular
aminoglycoside.
c) Decreased efficiency of the aminoglycoside transporting
mechanism: either the pores in the outer coat become less permeable or the
active transport is interfered. This again is not frequently encountered in the
clinical setting. In some Pseudomonas which
develop resistance, the antibiotic
induced 2nd phase active transport has been found to be deficient.
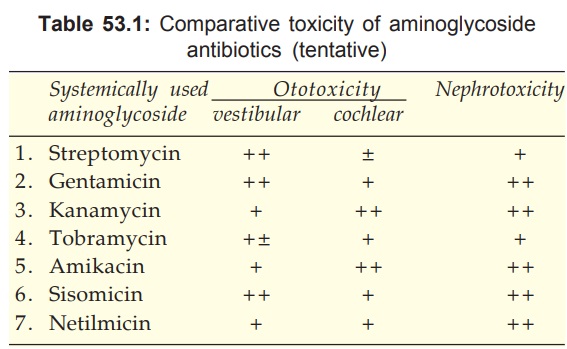
Shared Toxicities
The aminoglycosides
produce toxic effects which are common to all members, but the relative
propensity differs (see Table 53.1).
1. Ototoxicity
This is the most
important dose and duration of
treatment related adverse effect. The vestibular or the cochlear part may be primarily
affected by a particular aminoglycoside. These drugs are concentrated in the
labyrinthine fluid and are slowly removed from it when the plasma concentration
falls. Ototoxicity is greater when plasma concentration of the drug is
persistently high and above a threshold value. The vestibular/ cochlear sensory
cells and hairs undergo concentration dependent destructive changes.
Aminoglycoside ear drops can cause ototoxicity when instilled in patients with
perforated eardrum; contraindicated in them.
Cochlear Damage It starts from the base and spreads to the apex; hearing loss affects the
high frequency sound first, then progressively encompasses the lower
frequencies. No regeneration of the sensory cells occurs; auditory nerve fibres
degenerate in a retrograde manner—deafness is permanent. Older patients and
those with preexisting hearing defect are more susceptible. Initially, the
cochlear toxicity is asymptomatic; can be detected only by audiometry. Tinnitus
then appears, followed by progressive hearing loss. On stopping the drug,
tinnitus disappears in 4–10 days, but frequency loss persists.
Vestibular Damage Headache is usually
first to appear, followed by
nausea, vomiting, dizziness, nystagmus, vertigo and ataxia. When the drug is
stopped at this stage, it passes into a chronic phase lasting 6 to 10 weeks in
which the patient is asymptomatic while in bed and has difficulty only during
walking. Compensation by visual and proprioceptive positioning and recovery
(often partial) occurs over 1–2 years. Permanency of changes depends on the
extent of initial damage and the age of the patient (elderly have poor recovery).
2. Nephrotoxicity
It manifests as
tubular damage resulting in
loss of urinary concentrating power, low g.f.r., nitrogen retention, albuminuria
and casts. Aminoglycosides attain high concentration in the renal cortex and
toxicity is related to the total amount of the drug received by the patient. It
is more in the elderly and in those with preexisting kidney disease.
Essentially, renal damage caused by aminoglycosides is totally reversible,
provided the drug is promptly discontinued. It has been suggested that aminoglycosides
interfere with the production of PGs in the kidney and that this is causally
related to the reduced g.f.r. An important implication of aminoglycoside-induced
nephrotoxicity is reduced clearance of the antibiotic → higher blood levels → enhanced ototoxicity.
3. Neuromuscular
Blockade
All aminoglycosides
reduce ACh release from the motor nerve endings: interfere with mobilization of
centrally located synaptic vesicles to fuse with the terminal membrane
(probably by antagonizing Ca2+) as well as decrease the sensitivity of the
muscle endplates to Ach. The effect of this action is not manifested ordinarily
in the clinical use of these drugs. However, apnoea and fatalities have
occurred when these antibiotics were put into peritoneal or pleural cavity after
an operation, especially if a curare-like muscle relaxant was administered
during surgery. Rapid absorption form the peritoneum/pleura produces high blood
levels and adds to the residual action of the neuromuscular blocker.
Neomycin and streptomycin have higher propensity than kanamycin,
gentamicin or amikacin; tobramycin is least likely to produce this effect. The neuromuscular
block can be partially antagonized by i.v. injection of a calcium salt.
Neostigmine has inconsistent reversing action.
Myasthenic weakness is accentuated by these drugs. Neuromuscular
blockers should be used cautiously in patients receiving aminoglycosides.
Precautions And Interactions
1.
Avoid aminoglycosides during pregnancy: risk
of foetal ototoxicity.
2.
Avoid concurrent use of other ototoxic drugs,
e.g. high ceiling diuretics, minocycline.
3. Avoid concurrent use of other nephrotoxic
drugs, e.g. amphotericin B, vancomycin, cyclosporine and cisplatin.
4.
Cautious use in patients past middle age and
in those with kidney damage.
5.
Cautious use of muscle relaxants in patients
receiving an aminoglycoside.
6.
Do not mix aminoglycoside with any drug in the
same syringe/infusion bottle.
STREPTOMYCIN
It is the oldest
aminoglycoside antibiotic obtained from Streptomyces
griseus; used extensively in the past, but now practically restricted to
treatment of tuberculosis. It is less potent (MICs are higher) than other aminoglycosides.
The antimicrobial spectrum of streptomycin is relatively narrow: active
primarily against aerobic gram-negative bacilli, but potency is low. Sensitive
organisms are—H. ducreyi, Brucella,
Yersinia pestis, Francisella
tularensis, Nocardia, Calym. granulomatis, M. tuberculosis. Only few
strains of E. coli, H. influenzae, V.
cholerae, Shigella, Klebsiella, enterococci and some gram-positive cocci are
now inhibited, that too at higher concentrations. All other organisms including
Pseudomonas are unaffected.
Resistance
Many organisms develop
rapid resistance to
streptomycin, either by onestep mutation or by acquisition of plasmid which
codes for inactivating enzymes. In the intestinal and urinary tracts, resistant
organisms may emerge within 2 days of therapy. E. coli., H. influenzae, Str. pneumoniae, Str. pyogenes, Staph. aureus have
become largely resistant. If it is used alone,
M. tuberculosis also become
resistant.
Streptomycin Dependence
Certain mutants grown
in the presence of
streptomycin become dependent on it. Their growth is promoted rather than
inhibited by the antibiotic. This occurs when the antibiotic induced misreading
of the genetic code becomes a normal feature for the organism. This phenomenon
is probably significant only for use of streptomycin in tuberculosis.
Cross Resistance
Only partial and often unidirectional cross resistance occurs between
streptomycin and other aminoglycosides.
Pharmacokinetics
Streptomycin is highly ionized. It is neither absorbed nor destroyed
in the g.i.t. However, absorption from injection site in muscles is rapid. It
is distributed only extracellularly: volume of distribution (0.3 L/kg) is
nearly equal to the extracellular fluid volume. Low concentrations are attained
in serous fluids like synovial, pleural, peritoneal. Concentrations in CSF and
aqueous humour are often nontherapeutic, even in the presence of inflammation.
Plasma protein binding is clinically insignificant.
Streptomycin is not
metabolized—excreted unchanged in urine. Glomerular filtration is the main
channel: tubular secretion and reabsorption are negligible. The plasma t½ is
2–4 hours, but the drug persists longer in tissues. Renal clearance of
streptomycin parallels creatinine clearance and is approximately 2/3 of it.
Halflife is prolonged and accumulation occurs in patients with renal
insufficiency, in the elderly and neonates who have low g.f.r. Reduction in
dose or increase in dose-interval is essential in these situations.
These pharmacokinetic
features apply to all systemically administered aminoglycosides.
Adverse Effects
About 1/5 patients
given streptomycin 1 g BD
i.m. experience vestibular disturbances. Auditory disturbances are less common.
Streptomycin has the lowest nephrotoxicity among aminoglycosides;
probably because it is not concentrated in the renal cortex. Hypersensitivity
reactions are rare; rashes, eosinophilia, fever and exfoliative dermatitis have
been noted. Anaphylaxis is very rare. Topical use is contraindicated for fear of
contact sensitization.
Superinfections are not significant. Pain at injection site is
common. Paraesthesias and scotoma are occasional.
AMBISTRYNS 0.75, 1 g
dry powder per vial for inj.
Acute infections: 1 g
(0.75 g in those above 50 yr age) i.m. BD for 7–10 days.
Tuberculosis: 1 g or 0.75 g i.m. OD or twice weekly for 30–60
days.
Uses
1. Tuberculosis: see Ch. No. 55.
2. Subacute bacterial endocarditis (SABE): Streptomycin (now
mostly gentamicin) is given in conjunction with penicillin. A 4–6 weeks
treatment is needed.
3. Plague: It effects rapid cure (in 7–12 days), may be employed
in confirmed cases, but tetracyclines have been more commonly used for mass
treatment of suspected cases during an epidemic.
4. Tularemia: Streptomycin is the drug of choice for this rare
disease: effects cure in 7–10 days. Tetracyclines are the alternative drugs,
especially in milder cases.
In most other situations, e.g. urinary tract infection,
peritonitis, septicaemias, etc. where streptomycin was used earlier, gentamicin
or one of the newer aminoglycosides is now preferred due to low potency and
widespread resistance to streptomycin.
Oral use of streptomycin
for diarrhoea is banned in India.
GENTAMICIN
It was obtained from Micromonospora
purpurea in 1964; has become the most commonly used aminoglycoside for
acute infections. The properties of gentamicin including plasma t½ of 2–4 hours
after i.m. injection are the same as described above for streptomycin, but
there are following differences:
a) It is more potent (MIC for most organisms is
4–8 times lower.)
b) It has a broader spectrum of action: effective
against Ps. aeruginosa and most strains
of Proteus, E. coli, Klebsiella, Enterobacter, Serratia.
c)
It is ineffective against M. tuberculosis, Strep. pyogenes
and Strep. pneumoniae, but
inhibits many Strep. faecalis and
some Staph. aureus.
d) It is relatively more nephrotoxic.
Dose: The dose of gentamicin must be precisely calculated according to body weight and level
of renal function. For an average adult with normal renal function (creatinine
clearance > 100 ml/ min) 3–5 mg/kg/day i.m. either as single dose or
divided in three 8 hourly doses is recommended.
Because of
concentration dependent bactericidal and post-antibiotic effect of aminoglycosides,
it was theorised that high plasma concentration attained after the single daily
dose will be more effective. It is also likely to be less ototoxic because plasma
concentrations will remain subthreshold for ototoxicity for a longer period each
day allowing washout of the drug from the endolymph. The efficacy and safety of
many aminoglycosides by the conventional (thrice daily) and once daily regimens
has been compared in several studies. The data indicate similar efficacy and a
trend towards less toxicity. As such, many hospitals now practice once daily
dosing of aminoglycosides. It is more convenient as well.
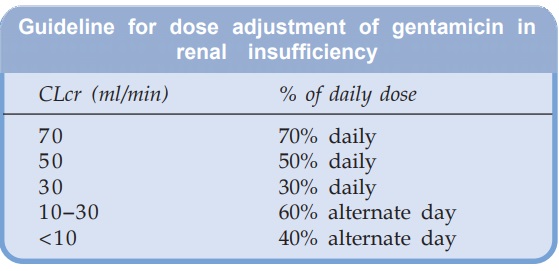
The daily dose of
gentamicin (and other aminoglycosides) should be reduced in patients with
impaired renal function according to measured creatinine clearance. A general
guideline is:
GARAMYCIN,
GENTASPORIN, GENTICYN 20, 60, 80, 240 mg per vial inj; also 0.3% eye/ear drops,
0.1% skin cream.
Uses
Gentamicin is the
cheapest (other than streptomycin) and the
first line aminoglycoside antibiotic. However, because of low therapeutic
index, its use should be restricted to serious gram-negative bacillary
infections.
1. Gentamicin is very valuable for preventing and treating
respiratory infections in critically ill patients; in those with impaired host
defence (receiving anticancer drugs or highdose corticosteroids; AIDS;
neutropenic), patients in resuscitation wards, with tracheostomy or on
respirators; postoperative pneumonias; patients with implants and in intensive
care units. It is often combined with a penicillin/cephalosporin or another
antibiotic in these situations. However, resistant strains have emerged in many
hospitals and nosocomial infections are less amenable to gentamicin now.
Another aminoglycoside (tobramycin, amikacin, sisomicin, netilmicin) is then
selected on the basis of the local sensitivity pattern. Aminoglycosides should
not be used to treat community acquired pneumonias caused by gram-positive
cocci and anaerobes.
Gentamicin is often added to the peritoneal dialysate to prevent
or treat peritonitis.
2. Pseudomonas,
Proteus or Klebsiella infections: burns, urinary tract infection,
pneumonia, lung abscesses, osteomyelitis, middle ear infection, septicaemia,
etc. are an important area of use of gentamicin. It may be combined with
piperacillin or a third generation cephalosporin for serious infections.
Topical use on infected burns and in conjunctivitis is permissible.
3. Meningitis caused by gram-negative bacilli: in addition to
the usual i.m. dose, 4 mg intrathecal injection may be given daily, but
benefits are uncertain. Because this is a serious condition, drug combinations
including an aminoglycoside are often used. The third generation cephalosporins
alone or with an aminoglycoside are favoured for this purpose.
4. SABE: gentamicin is more commonly used in place of
streptomycin to accompany penicillin.
Gentamicin-PMMA
(polymethyl methacrylate) chains (SEPTOPAL) is a special drug
delivery system for use in osteomyelitis. It consists of small acrylic
beads each impregnated with 7.5 mg gentamicin sulph. and threaded over surgical
grade wire. Implanted in the bone cavity after thorough removal of sequestra
and left in place for 10 days, it has improved cure rates.
KANAMYCIN
Obtained from S. kanamyceticus (in 1957), it was the
second systemically used aminoglycoside to be developed after streptomycin. It
is similar to streptomycin in all respects including efficacy against M. tuberculosis and lack of activity on Pseudomonas. However, it is more toxic,
both to the cochlea and to kidney. Hearing loss is more common than vestibular
disturbance.
Because of toxicity
and narrow spectrum of activity, it has been largely replaced by other
aminoglycosides for treatment of gram-negative bacillary infections. It is occasionally
used as a second line drug in resistant tuberculosis.
Dose: 0.5 g i.m. BD–TDS: KANAMYCIN, KANCIN, KANAMAC 0.5, 1 g inj.
TOBRAMYCIN
It was obtained from S. tenebrarius in the 1970s. The
antibacterial and pharmacokinetic properties, as well as dosage are almost identical
to gentamicin, but it is 2–4 times more active against Pseudomonas and Proteus,
including those resistant to gentamicin. However, it is not useful for
combining with penicillin in the treatment of enterococcal endocarditis. It
should be used only as a reserve alternative to gentamicin. Serious infections
caused by Pseudomonas and Proteus are its major indications.
Ototoxicity and nephrotoxicity is probably lower than gentamicin.
Dose: 3–5 mg/kg day in 1–3
doses.
TOBACIN 20, 60, 80 mg
in 2 ml inj. 0.3% eye drops. TOBRANEG 20, 40, 80 mg per 2 ml inj, TOBRABACT 0.3%
eye drops.
AMIKACIN
It is a semisynthetic
derivative of kanamycin to which it resembles in pharmacokinetics, dose and
toxicity. The outstanding feature of amikacin is its resistance to bacterial
aminoglycoside inactivating enzymes. Thus, it has the widest spectrum of
activity, including many organisms resistant to other aminoglycosides. However,
relatively higher doses are needed for Pseudomonas,
Proteus and Staph. infections.
The range of
conditions in which amikacin can be used is the same as for gentamicin. It is
recommended as a reserve drug for hospital acquired gram-negative bacillary
infections where gentamicin/tobramycin resistance is high. It is effective in
tuberculosis, but rarely used for this purpose. More hearing loss than
vestibular disturbance occurs in toxicity.
Dose: 15 mg/kg/day in 1–3
doses; urinary tract infection 7.5
mg/kg/day.
AMICIN, MIKACIN,
MIKAJECT 100 mg, 250 mg, 500 mg in 2 ml inj.
SISOMICIN
Introduced in 1980s,
it is a natural aminoglycoside from Micromonospora
inyoensis that is chemically and pharmacokinetically similar to gentamicin,
but somewhat more potent on Pseudomonas,
a few other gram-negative bacilli and β haemolytic Streptococci. It is moderately active on faecal Streptococci—can be combined with penicillin for SABE. However, it is susceptible to
aminoglycoside inactivating enzymes and offers no advantage in terms of
ototoxicity and nephrotoxicity. It can be used interchangeably with gentamicin
for the same purposes in the same doses.
ENSAMYCIN, SISOPTIN 50 mg, 10 mg (pediatric) per ml in 1 ml
amps, 0.3% eyedrops, 0.1% cream.
NETILMICIN
This semisynthetic
derivative of sisomicin has a broader spectrum of activity than gentamicin. It
is relatively resistant to aminoglycoside inactivating enzymes and thus
effective against many gentamicin-resistant strains. It is more active against Klebsiella, Enterobacter and Staphylococci, but less active against Ps. aeruginosa.
Pharmacokinetic characteristics and dosage of netilmicin are
similar to gentamicin. Experimental studies have shown it to be less ototoxic
than gentamicin and tobramycin, but clinical evidence is inconclusive: hearing
loss occurs, though fewer cases of vestibular damage have been reported.
A marginal improvement in antibacterial spectrum, clinical
efficacy and possibly reduced toxicity indicates that netilmicin could be
preferable in critically ill and neutropenic patients, and retain activity in
hospitals where gentamicin resistance has spread.
Dose: 4–6 mg/kg/day in 1–3
doses; NETROMYCIN 10, 25, 50 mg in 1 ml, 200 mg in 2 ml and 300 mg
in 3 ml inj., NETICIN 200 mg (2 ml), 300 mg (3 ml) inj.
NEOMYCIN
Obtained from S. fradiae , it is a widespectrum
aminoglycoside, active against most gram-negative bacilli and some gram-positive
cocci.
However, Pseudomonas and Strep. pyogenes are not sensitive. Neomycin is highly toxic to the
internal ear (mainly auditory) and to kidney. It is, therefore, not used systemically.
Absorption from the g.i.t. is minimal. Oral and topical administration does not
ordinarily cause systemic toxicity.
Dose: 0.25–1 g QID oral,
0.3–0.5% topical.
NEOMYCIN SULPHATE 350,
500 mg tab, 0.3% skin oint, 0.5% skin cream, eye oint.
NEBASULF: Neomycin
sulph. 5 mg, bacitracin 250 U, sulfacetamide 60 mg/g oint. and powder for
surface application.
POLYBIOTIC CREAM:
Neomycin sulph. 5 mg, polymyxin 5,000 IU, gramicidin 0.25 mg/g cream.
NEOSPORIN: Neomycin
3400 iu, polymyxin B 5000 iu, bacitracin 400 iu/g oint and powder for surface application.
NEOSPORINH: Neomycin 3400 iu, polymyxin B 5000 iu, hydrocortisone 10 mg per g
oint and per ml ear drops.
Uses
1. Topically (often in
combination with polymyxin, bacitracin, etc.) for infected wound, ulcers, burn,
external ear infections, conjunctivitis, but like other topical antiinfective
preparations, benefits are limited.
2. Orally for:
a) Preparation of
bowel before surgery: (3 doses of 1.0 g along with metronidazole 0.5 g on day
before surgery) may reduce postoperative infections.
b) Hepatic coma: Normally
NH3 is produced by colonic bacteria. This is absorbed and converted
to urea by liver. In severe hepatic failure, detoxication of NH3
does not occur, blood NH3 levels rise and produce encephalopathy. Neomycin,
by suppressing intestinal flora, diminishes NH3 production and
lowers its blood level; clinical improvement is seen within 2–3 days. However,
because of toxic potential it is infrequently used for this purpose; lactulose is
preferred.
Adverse Effects
Applied topically neomycin has low sensitizing potential. However, rashes
do occur.
Oral neomycin has a
damaging effect on intestinal villi—prolonged treatment can induce malabsorption
syndrome with diarrhoea and steatorrhoea. It can decrease the absorption of
digoxin and many other drugs, as well as bile acids.
Due to marked
suppression of gut flora, superinfection by Candida
can occur.
Small amounts that are
absorbed from the gut or topical sites are excreted unchanged by kidney. This may
accumulate in patients with renal insufficiency—cause further kidney damage and
ototoxicity. Neomycin is contraindicated if renal function is impaired.
Applied to serous
cavities (peritoneum), it can cause apnoea due to muscle paralysing action. Neomycin
containing anti-diarrhoeal formulations are banned in India.
FRAMYCETIN
Obtained from S. lavendulae, it is very similar to
neomycin. It is too toxic for systemic administration and is used topically on
skin, eye, ear in the same manner as neomycin.
SOFRAMYCIN, FRAMYGEN
1% skin cream, 0.5% eye drops or oint.
