Factors Modifying Drug Action
| Home | | Pharmacology |Chapter: Essential pharmacology : Aspects Of Pharmacotherapy; Clinical Pharmacology And Drug Development
Variation in response to the same dose of a drug between different patients and even in the same patient on different occasions is a rule rather than exception. One or more of the following categories of differences among individuals are responsible for the variations in drug response:
FACTORS MODIFYING DRUG ACTION
Variation
in response to the same dose of a drug between different patients and even in
the same patient on different occasions is a rule rather than exception. One or
more of the following categories of differences among individuals are responsible
for the variations in drug response:
§ Individuals differ in
pharmacokinetic handling of drugs: attain varying plasma/target site concentration
of the drug. This is more marked for drugs disposed by metabolism (e.g. propranolol)
than for drugs excreted unchanged (e.g. atenolol).
§ Variations in number
or state of receptors, coupling proteins or other components of response
effectuation.
§ Variations in neurogenic/hormonal
tone or concentrations of specific constituents, e.g. atropine tachycardia
depends on vagal tone, propranolol bradycardia depends on sympathetic tone,
captopril hypotension depends on body Na+ status.
A multitude of host and
external factors influence drug response. They fall in two categories viz genetic and nongenetic including all environmental, circumstantial and personal
variables. Though individual variation cannot be totally accounted for by these
factors, their understanding can guide the choice of appropriate drug and dose
for an individual patient. be totally accounted for by these factors, their
understanding can guide the choice of appropriate drug and dose for an
individual patient. However, final adjustments have to be made by observing the
response in a given patient on a given occasion.
The factors modify
drug action either:
a) Quantitatively The plasma
concentration and/or the action of
the drug is increased or decreased. Most of the factors introduce this type of
change and can be dealt with by adjustment of drug dosage.
b) Qualitatively The type of response
is altered, e.g. drug allergy or
idiosyncrasy. This is less common but often precludes further use of that drug
in the affected patient.
The various factors
are discussed below—
1. Body Size
It influences the concentration of the drug attained at
the site of action. The average adult dose refers to individuals of medium
built. For exceptionally obese or lean individuals and for children dose may be
calculated on body weight (BW) basis:
BW (kg)
Individual dose = ————
× average adult dose
70
It has been argued
that body surface area (BSA) provides a more accurate basis for dose calculation,
because total body water, extracellular fluid volume and metabolic activity are
better paralleled by BSA.
BSA (m2 )
Individual dose = ————
× average adult dose
1.7
The BSA of an individual
can be calculated from Dubois formula:
BSA (m2) = BW
(kg)0.425 × Height (cm)0.725 × 0.007184
or obtained from chartform
or sliderule nomograms based on BW and height.
However, dose
recommendations in terms of BSA are available only for anticancer and a handful
of other drugs: for the rest BW has been used as the index. Thus, prescribing
on BSA basis suffers from lack of data base, is more cumbersome and has not
thrived, except in few cases.
2. Age
The dose of a drug for children is often calculated from the
adult dose
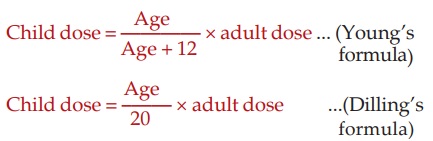
It can also be
calculated (more accurately) on BW or BSA basis (see above), and for many drugs, manufacturers give dosage
recommendations on mg/kg basis. Average figures for children are given below.
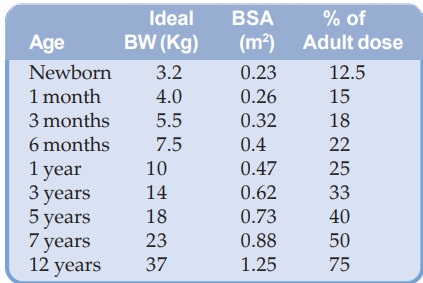
However, infants and children are not small adults. They have important physiological
differences from adults. The newborn
has low g.f.r. and tubular transport is immature. As such, the t½ of drugs
excreted by glomerular filtration (gentamicin) and tubular secretion
(penicillin) is prolonged by 3 to 5 times. Glomerular filtration reaches adult
rates by 5 month of age and tubular secretion takes about 7 months to mature.
Similarly, hepatic drug metabolizing system is inadequate in newborns
—chloramphenicol can produce gray baby
syndrome. Bloodbrain barrier is more permeable—drugs attain higher concentration
in the CNS (accumulation of unconjugated bilirubin causes kernicterus). These defects are exaggerated in the premature infant.
Drug absorption may also be altered in infants because of lower gastric acidity
and slower intestinal transit. Transdermal absorption however, is faster
because their skin is thin and more permeable. Therefore, infant doses must be learned
as such and not derived from any formula.
After
the first year of life, drug metabolism is often faster than in adults, e.g.
theophylline, phenytoin, carbamazepine t½ is shorter in children. Also, higher
per kg dose is needed for drugs which are primarily excreted unchanged by
kidney, e.g. daily dose of digoxin is about 8–12 μg/kg compared to adult
dose of 3–5
μg/kg.
Solid
dosage forms and aerosol inhalations are difficult to administer to young
children.
Children are growing
and are susceptible to special adverse effects of drugs, e.g. suppression of
growth can occur with corticosteroids; androgens may promote early fusion of
epiphysis resulting in stunting of stature; tetracyclines get deposited in
growing teeth and discolour/deform them. Dystonic reactions to phenothiazines
are more common in children.
Elderly In the elderly, renal
function progressively declines (intact nephron loss) so that g.f.r. is ~ 75%
at 50 years and ~ 50% at 75 years age compared to young adults. Drug doses have
to be reduced, e.g. daily dose of streptomycin is 0.75 g after 50 years and 0.5
g after 70 years of age compared to 1 g for young adults. There is also a reduction
in the hepatic microsomal drug metabolizing activity and liver blood flow: oral
bioavailability of drugs with high hepatic extraction is generally increased, but
the overall effects on drug metabolism are not uniform. Due to lower renal as
well as metabolic clearance, the elderly are prone to develop cumulative
toxicity while receiving prolonged medication. Other affected aspects of drug
handling are slower absorption due to reduced motility of and blood flow to
intestines, lesser plasma protein binding due to lower plasma albumin,
increased or decreased volume of distribution of lipophilic and hydrophilic
drugs respectively. Aged are relatively intolerant to digitalis. The responsiveness
of adrenergic receptors to both agonists and antagonists is reduced in the
elderly and sensitivity to other drugs also may be altered. Due to prostatism
in elderly males, even mild anticholinergic activity of the drug can accentuate
bladder voiding difficulty. Elderly are also likely to be on multiple drug
therapy for hypertension, ischaemic heart disease, diabetes, arthritis, etc.
which increases many fold the chances of drug interactions. They are more prone
to develop postural instability, giddiness and mental confusion. In general,
the incidence of adverse drug reactions is much higher in the elderly.
3. Sex
Females have smaller
body size and require doses that are
on the lower side of the range. Subjective effects of drugs may differ in females
because of their mental makeup. Maintenance treatment of heart failure with
digoxin is reported to be associated with higher mortality among women than
among men. A number of antihypertensives (clonidine, methyldopa, βblockers, diuretics)
interfere with sexual function in males but not in females. Gynaecomastia is a
side effect (of ketoconazole, metoclopramide, chlorpromazine, digitalis) that
can occur only in men. Ketoconazole causes loss of libido in men but not in women.
Obviously androgens are unacceptable to women and estrogens to men. In women
consideration must also be given to menstruation, pregnancy and lactation.
Drugs given during pregnancy can affect the foetus. There are
marked and progressive physiological changes during pregnancy, especially in
the third trimester, which can alter drug disposition.
§
Gastrointestinal motility is reduced → delayed absorption of
orally administered drug.
§
Plasma and extracellular fluid volume
expands—volume of drug distribution may increase.
§
While plasma albumin level falls, that of α1 acid glycoprotein
increases—the unbound fraction of acidic drugs increases but that of basic
drugs decreases.
§
Renal blood flow increases markedly— polar
drugs are eliminated faster.
§
Hepatic
microsomal enzymes undergo induction—many drugs are metabolized
faster.
Thus, the overall effect on drug disposition is complex and often
difficult to predict.
4. Species
and race
There are many examples of differences in responsiveness to
drugs among different species; rabbits are resistant to atropine, rats and mice
are resistant to digitalis and rat is more sensitive to curare than cat. These
differences are important while extrapolating results from experimental animals
to man.
Among human beings
some racial differences have been observed, e.g. blacks require higher and mongols
require lower concentrations of atropine and ephedrine to dilate their pupil. βblockers are less
effective as antihypertensive in AfroCaribbeans. Indians tolerate thiacetazone
better than whites. Considering the widespread use of chloramphenicol in India
and Hong Kong, relatively few cases of aplastic anaemia have been reported compared
to its incidence in the west. Similarly, quiniodochlor related cases of
subacute myelooptic neuropathy (SMON)
occurred in epidemic
proportion in Japan, but there is no confirmed report of its occurrence in
India despite extensive use.
5. Genetics
The dose of a drug to produce the same effect may vary
by 4–6 fold among different individuals. All key determinants of drug response,
viz. transporters, metabolizing
enzymes, ion channels, receptors with their couplers and effectors are
controlled genetically. Hence, a great deal of individual variability can be
traced to the genetic composition of the subject. The study of genetic basis
for variability in drug response is called ‘Pharmacogenetics’.
It deals with genetic influences on drug action as well as on drug handling by
the body. As the genomic technology has advanced, gene libraries and huge data
bases (like ‘pharmacogenetics and pharmacogenomics knowledge base’, ‘Human
genome variation database’, etc.) have been created aiming at improving
precision in drug therapy.
Pharmacogenomics is the use of genetic information to guide the choice of drug
and dose on an individual basis. It intends to identify individuals who are
either more likely or less likely to respond to a drug, as well as those who
require altered dose of certain drugs. Attempt is made to define the genetic
basis of an individual’s profile of drug response and to predict the best
treatment option for him/her. So far, this has been applied largely to patients
with known genetic abnormalities, but the goal is ‘personalized medicine’ on a
wide scale. However, a large proportion of genetic variability still remains
unaccounted for.
A
continuous variation with Gaussian frequency distribution is seen in the case
of most drugs. In addition, there are some specific genetic defects which lead
to discontinuous variation in drug responses, e.g.—
§ Atypical pseudocholinesterase
results in prolonged succinylcholine apnoea.
§ G6PD deficiency is
responsible for haemolysis with primaquine and other oxidizing drugs like
sulfonamides, dapsone, quinine, nalidixic acid, nitrofurantoin and menadione,
etc.
§ The low activity CYP2C9
variants metabolize warfarin at a slow rate and are at higher risk of bleeding.
§ Thiopurine methyl
transferase (TPMT) deficiency increases risk of severe bone marrow toxicity of
6mercaptopurine and azathioprine.
§ Irinotecan induced
neutropenia and diarrhoea is more in patients with UGT1A1 *28 allele of
glucuronyl transferase.
§ Severe 5fluorouracil
toxicity occurs in patients with dihydropyrimidine dehydrogenase (DPD)
deficiency.
§ Over expression of Pgp
results in tumour resistance to many cancer chemotherapeutic drugs, because it
pumps out the drug from the tumour cells.
§ Polymorphism of Nacetyl
transferase 2 (NAT2) gene results in rapid and slow acetylator status.
Isoniazid neuropathy, procainamide and hydralazine induced lupus occurs mainly
in slow acetylators.
§ Acute intermittent
porphyria—precipitated by barbiturates is due to genetic defect in repression
of porphyrin synthesis.
§ CYP2D6 abnormality
causes poor metoprolol/ debrisoquin metabolizer status. Since several
antidepressants and antipsychotics also are substrates of CYP2D6, deficient
patients are more likely to experience their toxicity. Codeine fails to produce
analgesia in CYP2D6 deficient, because this enzyme generates morphine from
codeine.
§ Malignant hyperthermia
after halothane is due to abnormal Ca2+ release channel (ryanodine
§ receptor) in the
sarcoplasmic reticulum of skeletal muscles.
§ Inability to hydroxylate
phenytoin results in toxicity at usual doses.
§ Resistance to coumarin
anticoagulants is due to an abnormal enzyme (that regenerates the reduced form
of vit. K) which has low affinity for the coumarins.
§ Attack of angle
closure glaucoma is precipitated by mydriatics in individuals with narrow
iridocorneal angle.
Genotype to phenotype
predictability is much better in monogenic phenotypic traits such as G6PD,
CYP2D6, TPMT, etc., than for multigenic traits. Majority of gene polymorphisms
are due to substitution of a single base pair by another. When found in the
population at a frequency of >1%, these are called ‘Single neucleotide polymorphisms’
(SNPs). Gene polymorphisms are often encountered at different frequencies among
different ethnic/geographical groups.
Despite
accumulation of considerable pharmacogenomic data and the fact that genotyping
of the individual needs to be done only once, its practical application in
routine patient care is at present limited due to prerequirement of multiple
drug specific genotypic screening. Simple spot tests for some, e.g. G6 PD
deficiency are currently in use.
6. Route Of
Administration
Route
of administration governs the speed and intensity of drug response. Parenteral
administration is often resorted to for more rapid, more pronounced and more
predictable drug action. A drug may have entirely different uses through
different routes, e.g. magnesium sulfate given orally causes purgation, applied
on sprained joints—decreases swelling, while intravenously it produces CNS depression
and hypotension.
7.
Environmental Factors And Time Of
Administration
Several environmental
factors affect drug responses.
Exposure to insecticides, carcinogens, tobacco smoke and consumption of
charcoal broiled meat are well known to induce drug metabolism. Type of diet
and temporal relation between drug ingestion and meals can alter drug
absorption, e.g. food interferes with absorption of ampicillin, but a fatty
meal enhances absorption of griseofulvin. Subjective effects of a drug may be
markedly influenced by the setup in which it is taken. Hypnotics taken at night
and in quiet, familiar surroundings may work more easily. It has been shown
that corticosteroids taken as a single morning dose cause less pituitary-adrenal
suppression.
8. Psychological Factor
Efficacy
of a drug can be affected by
patient’s beliefs, attitudes and expectations. This is particularly applicable
to centrally acting drugs, e.g. a nervous and anxious patient requires more
general anaesthetic; alcohol generally impairs performance but if punishment
(which induces anxiety) is introduced, it may actually improve performance.
Placebo This is an inert substance which is given in the garb of a medicine. It works by
psychological rather than pharmacological means and often produces responses
equivalent to the active drug. Some individuals are more suggestible and easily
respond to a placebo— ‘placebo reactors’. Placebos are used in two situations:
§ As a control device in
clinical trial of drugs (dummy medication).
§ To treat a patient who,
in the opinion of the physician, does not require an active drug.
Placebo
is a Latin word meaning ‘I shall please’. A patient responds to the whole
therapeutic setting; placebo effect largely depends on the physician-patient
relationship.
Placebos
do induce physiological responses, e.g. they can release endorphins in brain—causing
analgesia. Naloxone, an opioid antagonist, blocks placebo analgesia. Placebo
effects can thus supplement pharmacological effects. However, placebo effects
are highly variable even in the same individual, e.g. a placebo may induce
sleep on the first night but not subsequently. Thus, it has a very limited role
in practical therapeutics. Substances commonly used as placebo are lactose
tablets/capsules and distilled water injection.
Nocebo It is the converse of placebo, and refers to negative psychodynamic effect evoked by loss
of faith in the medication and/or the physician. Nocebo effect can oppose the
therapeutic effect of active medication.
9. Pathological states
Not only drugs modify disease processes, several diseases can
influence drug disposition and drug action:
Gastrointestinal diseases These can alter absorption of orally administered drugs. The
changes are complex and drug absorption can increase or decrease, e.g. in coeliac
disease absorption of amoxicillin is decreased but that of cephalexin and
cotrimoxazole is increased. Thus, malabsorption syndrome does not necessarily
reduce absorption of all drugs. Gastric stasis occurring during migraine attack
retards the absorption of ingested drugs. Achlorhydria decreases aspirin
absorption by favouring its ionization. NSAIDs can aggravate peptic ulcer
disease.
Liver disease Liver disease
(especially cirrhosis) can influence drug disposition in several ways:
§
Bioavailability of drugs having high first
pass metabolism is increased due to loss of hepatocellular function and portocaval
shunting.
§
Serum albumin is reduced—protein binding of
acidic drugs (diclofenac, warfarin, etc.) is reduced and more drug is present
in the free form.
§
Metabolism and elimination of some drugs
(morphine, lidocaine, propranolol) is decreased—their dose should be reduced.
Alternative drugs that do not depend on hepatic metabolism for elimination
and/or have shorter t½ should be preferred, e.g. oxazepam or lorazepam in place
of diazepam; atenolol as βblocker.
§
Prodrugs needing hepatic metabolism for activation,
e.g. prednisone, bacampicillin, sulindac are less effective and should be avoided.
The
changes are complex and there is no simple test (like creatinine clearance for
renal disease) to guide the extent of alteration in drug disposition; kinetics
of different drugs is affected to different extents.
Drug
action as well can be altered in liver disease in the case of certain drugs,
e.g.
§ The sensitivity of
brain to depressant action of morphine and barbiturates is markedly increased
in cirrhotics—normal doses can produce coma.
§ Brisk diuresis can
precipitate mental changes in patients with impending hepatic encephalopathy,
because diuretics cause hypokalemic alkalosis which favours conversion of NH+4
to NH3 → enters brain more easily.
§ Oral anticoagulants
can markedly increase prothrombin time, because clotting factors are already
low.
§ Fluid retaining action
of phenylbutazone (also other NSAIDs) and lactic acidosis due to metformin are
accentuated.
Hepatotoxic
drugs should be avoided in liver disease.
Kidney disease It markedly affects
pharmacokinetics of many drugs as well as alters the effects of some drugs.
Clearance of drugs
that are primarily excreted unchanged (aminoglycosides, digoxin, phenobarbitone)
is reduced parallel to decrease in creatinine clearance (CLcr). Loading dose of such a drug is not altered (unless edema is
present), but maintenance doses should be reduced or dose interval prolonged
proportionately. A rough guideline is given in the box:
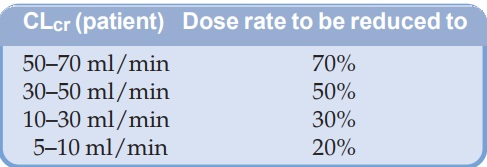
Dose
rate of drugs only partly excreted unchanged in urine also needs reduction, but
to lesser extents. If the t½ of the drug is prolonged, attainment of steady state
plasma concentration with maintenance doses is delayed proportionately.
Plasma
proteins, specially albumin, are often low or altered in structure in patients
with renal disease—binding of acidic drugs is reduced, but that of basic drugs
is not much affected.
The
permeability of blood-brain barrier is increased in renal failure; opiates,
barbiturates, phenothiazines, benzodiazepines, etc. produce more CNS
depression. Pethidine should be avoided because its metabolite norpethidine can
accumulate on repeated dosing and cause seizures. The target organ sensitivity
may also be increased. Antihypertensive drugs produce more postural hypotension
in patients with renal insufficiency.
Certain drugs worsen
the existing clinical condition in renal failure, e.g.
§ Tetracyclines have an
antianabolic effect and accentuate uraemia.
§ NSAIDs cause more
fluid retention.
§ Potentially
nephrotoxic drugs, e.g. cephalothin, aminoglycosides, tetracyclines (except
doxycycline), sulfonamides (crystalluria), vancomycin, cyclosporine,
amphotericin B should be avoided.
Antimicrobials needing
dose reduction in renal failure
Even in mild failure Only in severe
failure
Aminoglycosides Cotrimoxazole
Cephalexin Carbenicillin
Ethambutol Cefotaxime
Vancomycin Norfloxacin
Amphotericin B Ciprofloxacin
Acyclovir Metronidazole
Thiazide
diuretics tend to reduce g.f.r.: are ineffective in renal failure and can
worsen uraemia; furosemide should be used. Potassium sparing diuretics are
contraindicated; can cause hyperkalemia → cardiac depression.
Repeated doses of pethidine are likely to cause muscle twitching and seizures
due to accumulation of its excitatory metabolite norpethidine.
Urinary antiseptics
like nalidixic acid, nitrofurantoin and methenamine mandelate fail to achieve
high concentration in urine and are likely to produce systemic toxicity.
Congestive Heart Failure It can alter drug kinetics
by—
§
Decreasing drug absorption from g.i.t. due to
mucosal edema and splanchnic vasoconstriction. A definite reduction in procainamide
and hydrochlorothiazide absorption has been documented.
§
Modifying volume of distribution which can
increase for some drugs due to expansion of extracellular fluid volume or
decrease for others as a result of decreased tissue perfusion—loading doses of
drugs like lidocaine and procainamide should be lowered.
§
Retarding drug elimination as a result of
decreased perfusion and congestion of liver, reduced glomerular filtration rate
and increased tubular reabsorption; dosing rate of drugs may need reduction, as
for lidocaine, procainamide, theophylline.
§
The decompensated heart is more sensitive to
digitalis.
Thyroid Disease
The hypothyroid patients are more sensitive to
digoxin, morphine and CNS depressants. Hyperthyroid patients are relatively
resistant to inotropic action but more prone to arrhythmic action of digoxin.
The clearance of digoxin is roughly proportional to thyroid function, but this
only partially accounts for the observed changes in sensitivity.
Other
examples of modification of drug response by pathological states are:
§ Antipyretics lower
body temperature only when it is raised (fever).
§ Thiazides induce more
marked diuresis in edematous patients.
§ Myocardial infarction
patients are more prone to adrenaline and digitalis induced cardiac
arrhythmias.
§ Myasthenics are very sensitive
to curare.
§ Schizophrenics
tolerate large doses of phenothiazines.
§ Head injury patients
are prone to go into respiratory failure with normal doses of morphine.
§ Atropine, imipramine,
furosemide can cause urinary retention in individuals with prostatic hypertrophy.
§ Hypnotics given to a
patient in severe pain may cause mental confusion and delirium.
§ Cotrimoxazole produces
a much higher incidence of adverse reactions in AIDS patients.
10. Other Drugs
Drugs can modify the response to each other by pharmacokinetic
or pharmacodynamic interaction between them. Many ways in which drugs can
interact have already been considered.
11. Cumulation
Any drug will cumulate in the body if rate of
administration is more than the rate of elimination. However, slowly eliminated
drugs are particularly liable to cause cumulative toxicity, e.g. prolonged use
of chloroquine causes retinal damage.
• Full loading dose of digoxin should not be given if patient
has received it within the past week.
• A course of emetine should not be repeated within 6 weeks.
12. Tolerance
It refers to the requirement of higher dose of a drug
to produce a given response. Loss of therapeutic efficacy (e.g. of
salfonylureas in type 2 diabetes), which is a form of tolerance, is often
called ‘refractoriness’. Tolerance is
a widely occurring adaptive biological phenomenon. Drug tolerance may be:
Natural The species/individual is inherently less sensitive to the drug, e.g. rabbits are
tolerant to atropine; black races are tolerant to mydriatics. Some individuals
in any population are hyporesponders to certain drugs, e.g. to β adrenergic blockers
or to alcohol.
Acquired This occurs by repeated use of a drug in an individual who was initially responsive.
Body
is capable of developing tolerance to most drugs but the phenomenon is very
easily recognized in the case of CNS depressants. An uninterrupted presence of
the drug in the body favours development of tolerance. However, significant
tolerance does not develop to atropine, digitalis, cocaine, sodium
nitroprusside, etc. Tolerance need not develop equally to all actions of a drug,
consequently therapeutic index of a drug may increase or decrease with
prolonged use, e.g.:
§ Tolerance develops to
sedative action of chlorpromazine but not to its antipsychotic action.
§ Tolerance occurs to
the sedative action of phenobarbitone but not as much to its antiepileptic
action.
§ Tolerance occurs to
analgesic and euphoric action of morphine, but not as much to its constipating
and miotic actions.
Cross tolerance It is the development
of tolerance to pharmacologically related drugs, e.g. alcoholics are relatively
tolerant to barbiturates and general anaesthetics. Closer the two drugs are,
more complete is the cross tolerance between them, e.g.— There is partial cross
tolerance between morphine and barbiturates but complete cross tolerance between
morphine and pethidine.
Mechanisms responsible for development of tolerance are incompletely understood.
However, tolerance may be:
§
Pharmacokinetic/drug disposition tolerance—the
effective concentration of the drug at the site of action is decreased, mostly
due to enhancement of drug elimination on chronic use, e.g. barbiturates,
carbamazepine, amphetamine.
§
Pharmacodynamic/cellular tolerance— drug
action is lessened; cells of the target organ become less responsive, e.g. morphine,
barbiturates, nitrates. This may be due to down regulation of receptors, or weakening of response
effectuation.
Tachyphylaxis (Tachyfast, phylaxisprotection) is rapid development of tolerance when doses
of a drug repeated in quick succession result in marked reduction in response.
This is usually seen with indirectly acting drugs, such as ephedrine, tyramine,
nicotine. These drugs act by releasing catecholamines in the body, synthesis of
which is unable to match the rate of release: stores get depleted. Other mechanisms
like slow dissociation of the drug from its receptor, desensitization/internalization
or down regulation of receptor, etc. and/or compensatory homeostatic
adaptation.
Drug resistance It refers to tolerance
of microorganisms to inhibitory action of antimicrobials, e.g. Staphylococci to penicillin.
