Imidazoles and Triazoles
| Home | | Pharmacology |Chapter: Essential pharmacology : Antifungal Drugs
Four imidazoles are entirely topical, while ketoconazole is used both orally and topically. Two triazoles fluconazole and itraconazole have largely replaced ketoconazole for systemic mycosis because of greater efficacy, longer t½, fewer side effects and drug interactions.
IMIDAZOLES AND TRIAZOLES
These are presently
the most extensively used antifungal drugs.
Four imidazoles are
entirely topical, while ketoconazole is used both orally and topically. Two triazoles
fluconazole and itraconazole have largely replaced ketoconazole for systemic
mycosis because of greater efficacy, longer t½, fewer side effects and drug
interactions.
The imidazoles and
triazoles have broad-spectrum antifungal activity covering dermatophytes, Candida, other fungi involved in deep
mycosis (except mucor), Nocardia,
some gram-positive and anaerobic bacteria, e.g. Staph. aureus, Strep.
faecalis, Bac. fragilis and
Leishmania.
The mechanism of
action of imidazoles and triazoles is the same. They inhibit the fungal cytochrome
P450 enzyme ‘lanosterol 14demethylase’ and thus impair ergosterol synthesis
leading to a cascade of membrane abnormalities in the fungus. The lower host
toxicity of triazoles compared to imidazoles has correlated with their lower
affinity for mammalian CYP450 enzymes and lesser propensity to inhibit mammalian
sterol synthesis. However, because they are active against certain bacteria as
well (which do not have ergosterol), other mechanisms of action also appear to
be involved.
Development of fungal resistance to azoles has been noted among Candida infecting advanced AIDS
patients, but has not so far posed significant clinical problem.
Clotrimazole
It is effective in the
topical treatment of tinea
infections like ringworm: 60–100% cure rates are reported with 2–4 weeks
application on a twice daily schedule. Athletes’ foot, otomycosis and
oral/cutaneous/vaginal candidiasis have responded in >80% cases. It is
particularly favoured for vaginitis because of a long lasting residual effect
after once daily application. A 7 day course is generally used. For oropharyngeal
candidiasis 10 mg troche of clotrimazole is allowed to dissolve in the mouth
3–4 times a day, or the lotion/gel is applied/ swirled in the mouth for as long
as possible. It is also effective in skin infections caused by Corynebacteria.
Clotrimazole is well tolerated by most patients. Local
irritation with stinging and burning sensation occurs in some. No systemic
toxicity is seen after topical use.
SURFAZ, CLOTRIN, CLODERM 1% lotion, cream, powder; 100 mg
vaginal tab. CANDID 1% cream, gel, lotion, powder.
Econazole
It is similar to clotrimazole; penetrates superficial
layers of the skin and is highly effective in dermatophytosis, otomycosis, oral
thrush, but is somewhat inferior to clotrimazole in vaginitis. No adverse
effects, except local irritation in few is reported.
ECONAZOLE 1% oint, 150 mg vaginal tab; ECODERM 1% cream.
Miconazole
It is a highly
efficacious (>90% cure rate) drug for tinea,
pityriasis versicolor, otomycosis, cutaneous and vulvovaginal candidiasis.
Because of its good penetrating power, it has been found effective, though
partially, even in onychomycosis; single application on skin acts for a few days.
Irritation after
cutaneous application is infrequent. No systemic adverse effects are seen.
However, a higher incidence of vaginal irritation is reported in comparison to
clotrimazole; even pelvic cramps have been experienced.
DAKTARIN 2% gel, 2% powder
and solution; GYNODAKTARIN 2% vaginal gel; ZOLE 2% oint, lotion, dusting powder
and spray, 1% ear drops, 100 mg vaginal ovules.
Oxiconazole
Another recently
marketed topical imidazole antifungal
effective in tinea and other dermatophytic infection, as well as vaginal
candidiasis. Local irritation can occur in some patients.
OXIZON, ZODERM:
oxiconazole 1% with benzoic acid 0.25% cream/lotion; apply topically once or
twice daily.
Ketoconazole (KTZ)
It is the first orally
effective broad-spectrum antifungal drug, useful in both dermatophytosis and
deep mycosis. The oral absorption of KTZ is facilitated by gastric acidity
because it is more soluble at lower pH. Hepatic metabolism is extensive;
metabolites are excreted in urine and faeces. Elimination of KTZ is dose
dependent: t½ varies from 1½ to 6 hours. Penetration in CSF is poor: not
effective in fungal meningitis. However, therapeutic concentrations are
attained in the skin and vaginal fluid.
In spite of relatively
short t½, a single daily dose is satisfactory in less severe cases. The usual
dose is 200 mg OD or BD; higher doses are sometimes required.
FUNGICIDE, NIZRAL,
FUNAZOLE, KETOVATE 200 mg tab.
FUNGINOC 2% oint, 2% shampoo
(for dandruff), KETOVATE 2% cream. NIZRAL 2% cream, 2% lotion; DANRUF 2%
shampoo, HYPHORAL 2% lotion.
Adverse Effects
Ketoconazole is much
less toxic than AMB, but more
side effects occur than with itraconazole or fluconazole, that have largely
replaced it for systemic use.
The most common side effects
are nausea and vomiting; can be reduced by giving the drug with meals. Others
are—loss of appetite, headache, paresthesia, rashes and hair loss.
Ketoconazole decreases androgen production from testes, and it
displaces testosterone from protein binding sites. Gynaecomastia, loss of hair
and libido, and oligozoospermia may be the manifestations. Menstrual irregularities
occur in some women due to suppression of estradiol synthesis.
A dose-dependent decrease in serum hydrocortisone due to
synthesis inhibition has also been noted, but without any clinical
manifestations in normal individuals.
Mild and asymptomatic elevation of serum transaminases occurs in
~5% patients, but serious hepatotoxicity is infrequent.
It is contraindicated in pregnant and nursing women.
Interactions
H2 blockers, proton pump
inhibitors and antacids decrease the oral absorption of KTZ by reducing gastric
acidity.
Rifampin, phenobarbitone, carbamazepine and phenytoin induce KTZ
metabolism and reduce its efficacy.
Ketoconazole inhibits cytochrome P450, especially CYP3A4, and
raises the blood levels of several drugs including:
Phenytoin
Digoxin
Diazepam
Cyclosporine
Haloperidol
Nifedipine and other
DHPs
Warfarin
HIV protease
inhibitors
Sulfonylureas
Statin hypolipidaemics
The dangerous interaction with terfenadine, astemizole and
cisapride resulting in polymorphic ventricular tachycardia due to excessive
rise in plasma levels of these drugs has resulted in withdrawal of these drugs
from the market in many countries.
Use
Orally administered KTZ is effective in dermatophytosis because it is
concentrated in the stratum corneum; is
an alternative to griseofulvin, but use is restricted due to potential adverse
effects.
Though effective in monilial vaginitis, oral therapy (for
5–7 days) with KTZ is reserved for recurrent cases or those not responding to
topical agents.
Systemic Mycosis: Administered orally, KTZ is effective in several types of systemic
mycosis, but itraconazole and fluconazole, being more active with fewer side
effects, have largely replaced it for these indications except for
considerations of cost.
KTZ is occasionally
used in dermal leishmaniasis and kala
azar.
Highdose KTZ has been
used in Cushing’s syndrome to decrease corticosteroid production.
Fluconazole
It is a water-soluble
triazole having a wider range
of activity than KTZ; indications include cryptococcal meningitis, systemic and
mucosal candidiasis in both normal and immunocompromised patients, coccidioidal
meningitis and histoplasmosis.
Fluconazole is 94%
absorbed; oral bioavailability is not affected by food or gastric pH. It is
primarily excreted unchanged in urine with a t½ of 25–30 hr. Fungicidal concentrations
are achieved in nails, vagina and saliva; penetration into brain and CSF is
good. Dose reduction is needed in renal impairment.
Adverse Effects
Fluconazole produces
few side effects: mostly
nausea, vomiting, abdominal pain, rash and headache.
Selectivity for fungal
cytochrome P450 is higher; unlike KTZ, it does not inhibit steroid synthesis in
man: antiandrogenic and other endocrine side effects have not occurred.
Elevation of hepatic
transaminase has been noted in AIDS patients.
It is not recommended
in pregnant and lactating mothers.
Interactions
Though it affects
hepatic drug metabolism to a lesser
extent than KTZ, increased plasma levels of phenytoin, astemizole, cisapride,
cyclosporine, warfarin, zidovudine and sulfonylureas have been observed. A few
cases of ventricular tachycardia have been reported when fluconazole was given
with cisapride. The same caution as with KTZ or itraconazole needs to be
applied in co-administering other drugs. H2 blockers and proton pump
inhibitors do not effect its absorption.
Use
Fluconazole can be administered
orally as well as i.v. (in
severe infections).
A single 150 mg oral dose can cure vaginal candidiasis with few
relapses.
Oral fluconazole (150 mg/day for 2 weeks) is highly effective in
oropharyngeal candidiasis, but is reserved for cases not responding to topical
antifungals.
Most tinea infections and cutaneous candidiasis can be treated
with 150 mg weekly fluconazole for 4 weeks, while tinea unguium requires weekly
treatment for up to 12 months.
For disseminated candidiasis, cryptococcal/ coccidioidal
meningitis and other systemic fungal infections the dose is 200–400 mg/day for
4–12 weeks or longer. It is the preferred drug for fungal meningitis, because
of good CSF penetration. Long-term fluconazole maintenance therapy is needed in
AIDS patients with fungal meningitis.
An eye drop is useful in fungal keratitis. Fluconazole is
ineffective in aspergillosis and
mucormycosis, and inferior to itraconazole for histoplasmosis,
blastomycosis and sporotrichosis.
SYSCAN, ZOCON, FORCAN, FLUZON 50, 100, 150, 200 mg caps, 200
mg/100 ml i.v. infusion. SYSCAN 0.3% eye drops.
Itraconazole
This newer orally
active triazole antifungal has a
broader spectrum of activity than KTZ or fluconazole; includes some moulds like
Aspergillus. It is fungistatic, but
effective in immunocompromised
patients. Steroid hormone synthesis inhibition is absent in itraconazole, and
serious hepatotoxicity is rare.
Oral absorption of
itraconazole is variable. It is enhanced by food and gastric acid. Itraconazole
is highly protein bound, has a large volume of distribution (10L/Kg),
accumulates in vaginal mucosa, skin and nails, but penetration into CSF is
poor. It is largely metabolized in liver by CYP3A4; an active metabolite is produced
which is excreted in faeces; t½ varies from 30–64 hours. Itraconazole is well
tolerated in doses below 200 mg/day. Gastric intolerance is significant at 400
mg/day. Dizziness, pruritus, headache and hypokalaemia are the other common side
effects. Unsteadiness and impotence are infrequent. Plasma transaminase may
rise transiently. However, antiandrogenic and other hormonal adverse effects
are not seen. Impaired left ventricular function has been worsened in some
patients.
Drug Interactions
Oral absorption of
itraconazole is reduced by antacids, H2 blockers and proton pump
inhibitors.
Rifampin,
phenobarbitone, phenytoin and carbamazepine induce itraconazole metabolism and
reduce its efficacy.
On the other hand,
clarithromycin and HIV protease inhibitors reduce the metabolism of
itraconazole and raise its blood levels.
Itraconazole inhibits CYP3A4; drug interaction profile is
similar to KTZ; ventricular arrhythmias have occurred with terfenadine,
astemizole, cisapride and class III antiarrhythmics. Phenytoin, digoxin, sulfonylureas,
statins, dihydropyridines, protease inhibitors, warfarin and cyclosporine
levels are also increased.
Uses
Itraconazole is the
preferred azole antifungal for most
systemic mycosis (see Table 57.1) that
are not associated with meningitis. It is superior to fluconazole for histoplasmosis,
blastomycosis, sporotrichosis and is the drug of choice for para-coccidioidomycosis
and chromomycosis. It also affords some relief in aspergillosis. A dose of 200
mg OD/BD with meals is used for 3 months or more.
Vaginal Candidiasis: 200 mg OD for 3 days:
as effective as intravaginal clotrimazole. Dermatophytosis: 100–200 mg OD for
7–15 days: more effective than griseofulvin, but less effective than
fluconazole.
Onychomycosis: 200 mg/day for 3
months. An intermittent pulse regimen of 200 mg BD for 1 week each month for 3
months is equally effective. Relapses have occurred after itraconazole therapy,
though it remains in the nail for few months after completion of the course.
SPORANOX, CANDITRAL, CANDISTAT, ITASPOR, FLUCOVER 100 mg cap.
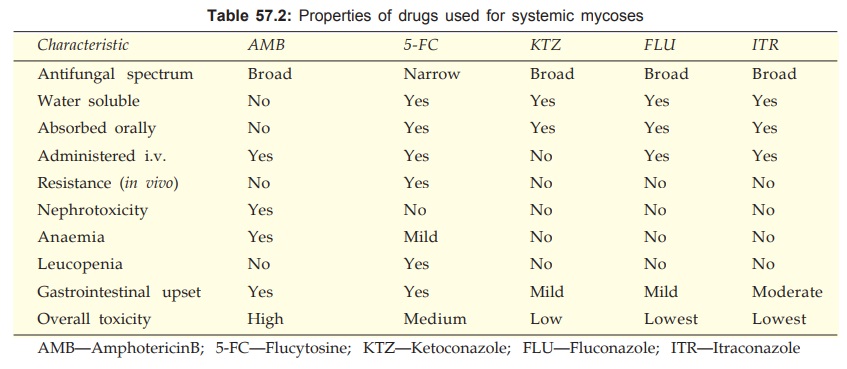
Important features of drugs used for systemic mycosis are
compared in Table 57.2.
Voriconazole
It is a second
generation broad-spectrum triazole introduced lately for difficult to treat
fungal infections like invasive aspergillosis, disseminated infections caused
by fluconazole resistant Candida,
Fusarium infections, and febrile neutropenia not responding to antibacterial
therapy. Serious cases are first treated i.v. followed by oral voriconazole. It
is metabolized by several CYP isoenzymes (CYP2C19, CYP3A4, etc) and inhibits
them as well. The drug interaction profile is similar to itraconazole. Rashes,
visual disturbances, QTc prolongation and an acute reaction on i.v. injection
are the significant adverse effects.
Terbinafine
This orally and topically
active drug against dermatophytes and Candida
belongs to a new allylamine class of antifungals. In contrast to azoles which
are primarily fungistatic, terbinafine is fungicidal: shorter courses of
therapy are required and relapse rates are low. It acts as a noncompetitive
inhibitor of ‘squalene epoxidase’, an early step enzyme in ergosterol
biosynthesis by fungi. Accumulation of squalene within fungal cells appears to
be responsible for the fungicidal action. The mammalian enzyme is inhibited
only by 1000fold higher concentration of terbinafine.
Approximately 75% of
oral terbinafine is absorbed, but only 5% or less from unbroken skin. First
pass metabolism further reduces oral bioavailability. It is lipophilic, widely
distributed in the body, strongly plasma protein bound and concentrated in
sebum, stratum corneum and nail plates. It is inactivated by metabolism and
excreted in urine (80%) and faeces (20%); elimination t½ of 11–16 hr is
prolonged to 10 days after repeated dosing.
Side effects of oral terbinafine
are gastric upset, rashes, taste disturbance. Some cases of hepatic
dysfunction, haematological disorder and severe cutaneous reaction are
reported. Enzyme inducers lower, and enzyme inhibitors raise its steady-state
plasma levels. Terbinafine does not inhibit CYP450.
Topical terbinafine
can cause erythema, itching, dryness, irritation, urticaria and rashes.
Use
Terbinafine applied
topically as 1% cream or orally 250 mg OD is
indicated in tinea pedis/ corporis/cruris/capitis and pityriasis versicolor; 2–6
weeks treatment is required according to the site. Onychomycosis is treated by
3–12 months oral therapy. Efficacy in toe nail infection is 60–80%, which is
higher than griseofulvin and itraconazole.
It is less effective
against cutaneous and mucosal candidiasis: 2–4 weeks oral therapy may be used
as an alternative to fluconazole.
LAMISIL, SEBIFIN,
DASKIL 250 mg tab, 1% topical cream. EXIFINE 125, 250 mg tabs, 1% cream; TERBIDERM
1% cream.
