Opioid Analgesics
| Home | | Pharmacology |Chapter: Essential pharmacology : Opioid Analgesics And Antagonists
A dark brown, resinous material obtained from poppy (Papaver somniferum) capsule. It contains two types of alkaloids.
OPIOID ANALGESICS
Opium
A dark brown, resinous
material obtained from poppy (Papaver
somniferum) capsule. It contains two types of alkaloids.
Phenanthrene Derivatives
Morphine (10% in
opium)
Codeine (0.5% in
opium)
Thebaine (0.2% in
opium), (Nonanalgesic)
Benzoisoquinoline Derivatives
Papaverine (1%) (Nonanalgesic)
Noscapine (6%) (Nonanalgesic)
Opium has been known
from the earliest times. It is mentioned in the Eber’s papyrus (1500 BC), in
the writings of Theophrastus (300 BC) and Galen (2nd century AD). Opium eating
became a social custom in China in the 18th century. Serturner, a pharmacist, isolated
the active principle of opium in 1806 and named it ‘morphine’ after the Greek god of dreams Morpheus. In the last century a large number of semisynthetic and
synthetic compounds have been developed with morphinelike, antagonistic and mixed
agonistic-antagonistic properties.
Morphine
Morphine is the
principal alkaloid in opium and still widely used. Therefore, it is described
as prototype.
Pharmacological Actions
1. CNS
Morphine has site
specific depressant and stimulant actions
in the CNS by interacting primarily with the μ opioid receptor as a
full agonist. The depressant actions are:
Analgesia Morphine is a strong
analgesic. Though dull, poorly
localized visceral pain is relieved better than sharply defined somatic pain;
higher doses can mitigate even severe pain—degree of analgesia increasing with
dose. Nociceptive pain arising from stimulation of peripheral pain receptors is
relieved better than neuretic pain (such as trigeminal neuralgia) due to inflammation
of or damage to neural structures. The associated reactions to intense pain
(apprehension, fear, autonomic effects) are also dampened. Suppression of pain
perception is selective, without affecting other sensations or producing
proportionate generalized CNS depression (contrast general anaesthetics).
Perception of pain and its emotional or suffering component are
both altered so that pain is no longer as unpleasant or distressing, i.e. the
patient tolerates pain better. The analgesic action of morphine has spinal and
supraspinal components. Intrathecal injection has been shown to cause segmental
analgesia without affecting other modalities. It acts in the substantia
gelatinosa of dorsal horn to inhibit release of excitatory transmitters from primary
afferents carrying pain impulses. The action appears to be exerted through
interneurones which are involved in the ‘gating’ of pain impulses. Release of
glutamate from primary pain afferents in the spinal cord and its postsynaptic
action on dorsal horn neurones is inhibited by morphine. Action at supraspinal
sites in medulla, midbrain, limbic and cortical areas may alter processing and
interpretation of pain impulses as well as send inhibitory impulses through
descending pathways to the spinal cord. Several aminergic and other neuronal
systems appear to be involved in the action of morphine. Simultaneous action at
spinal and supraspinal sites greatly amplifies the analgesia.
Sedation which is different
from that produced by hypnotics
is seen. Drowsiness and indifference to surroundings as well as to own body
occurs without motor incoordination, ataxia or apparent excitement (contrast
alcohol). Higher doses progressively induce sleep and coma. Morphine has no
anticonvulsant action, rather, fits may be precipitated.
Mood and Subjective Effects These are prominent.
Morphine has a calming effect; there is loss of apprehension, feeling of
detachment, lack of initiative, limbs feel heavy and body warm, mental clouding
and inability to concentrate occurs. In the absence of pain or apprehension,
these are generally appreciated as unpleasant by normal people. However,
patients in pain or anxiety, and especially addicts, perceive it as pleasurable
floating sensation: refer it as ‘high’. Rapid i.v. injection by addicts gives them
a ‘kick’ or ‘rush’ which is intensely pleasurable—akin to orgasm. Thus, one has
to learn to perceive the euphoric effect
of morphine.
The pleasurable and reinforcing effects of μ opioid agonists
(morphinelike) appear to involve a separate set of neuronal mechanisms than
those involved in analgesia and sedation. The euphoric effects are most likely
mediated by DA release in nucleus accumbance, whereas agonists (nalorphine
like) inhibit DA release and produce aversion. Inhibition of NA release in locus
ceruleus by opioids is implicated in their action to allay apprehension and
fear.
Respiratory Centre Morphine depresses respiratory
centre in a dose dependent manner; rate and tidal volume are both decreased:
death in poisoning is due to respiratory failure. Neurogenic, hypercapnoeic and
later hypoxic drives to the respiratory centre are suppressed in succession. In
addition, there is indifference to breathing: apnoeic patient may breath if
commanded.
Cough
centre It is depressed; more sensitive to morphine than
respiratory centre.
Temperature regulating centre It is depressed;
hypothermia occurs in cold surroundings.
Vasomotor centre It is depressed at
higher doses and contributes
to the fall in BP.
Morphine Stimulates:
a)
CTZ Nausea and vomiting occur
as side effects, especially if
stomach is full and the patient stands or moves about. Thus, morphine appears
to sensitize the CTZ to vestibular and other impulses. Larger doses depress
vomiting centre directly: emetics should not be tried in morphine poisoning.
b) Edinger Westphal Nucleus of III nerve is stimulated
producing miosis. This is a central action; no miosis occurs on topical
application of morphine to the eye. Mydriasis occurs in some species like cats.
Other ocular effect is a decrease in intraocular tension.
c)
Vagal centre It is stimulated→ bradycardia is the usual response.
d) Certain Cortical Areas and Hippocampal cells are stimulated.
Excitation is seen in an occasional individual. Muscular rigidity and
immobility is consistently manifested at high doses (especially on i.v.
injection): resembles catalepsy seen in rats and mice. Convulsions may occur in
morphine poisoning. The proconvulsant action has been ascribed to inhibition of
GABA release by hippocampal interneurones. Species like cat, lion, horse, sheep
and cow are uniformly excited and show hyperthermia.
2. Neuroendocrine
Hypothalamic
activation by afferent
collaterals is dampened. Hypothalamic influence on pituitary is reduced. As a
result FSH, LH, ACTH levels are lowered, while prolactin and GH levels are
raised (these are under predominant inhibitory control). The sex hormone and
corticosteroid levels are lowered in the short term, but tolerance develops in
the long term. Only few chronic abusers suffer from infertility; hypocorticism
is not a problem in them. Morphine can release ADH and reduce urine volume.
3. CVS
Morphine causes
vasodilatation due to:
(a) histamine release.
(b) depression of
vasomotor centre.
(c) direct action
decreasing tone of blood vessels. There is a shift of blood from pulmonary to
systemic circuit due to greater vasodilatation in the latter. Therapeutic doses
cause little change in the BP of recumbent normovolaemic patient. Postural
hypotension and fainting do occur due to impairment of vascular reflexes. Morphine
has little direct effect
on heart; rate
generally decreases due to stimulation of vagal centre, but may increase
reflexly if the BP falls. Cardiac work is consistently reduced due to decrease
in peripheral resistance. Intracranial tension tends to rise as a consequence
of CO2 retention leading to cerebral vasodilatation.
4. GIT
Constipation is a
prominent feature of morphine action. Several
factors contribute:
(a) Action directly on
intestines and in CNS increases tone and segmentation but decreases propulsive
movements. Tone of duodenum and colon may be increased to the level of spasm.
(b) Spasm of pyloric,
ileocaecal and anal sphincters.
(c) Decrease in all
gastrointestinal secretions: reduction in transfer of water and electrolytes
from mucosa to the lumen. Absorption of fluid is increased due to stasis.
(d) Central action
causing inattention to defecation reflex.
No tolerance develops
to this action: addicts remain chronically constipated.
5. Other Smooth
Muscles
Biliary tract
Morphine causes spasm of sphincter of Oddi → intrabiliary pressure
is increased → may cause biliary
colic. This action is only partly counteracted by atropine but more completely
by opioid antagonist naloxone and direct smooth muscle relaxants like nitrates.
Urinary
bladder Tone of both detrusor and sphincter is increased
→ urinary urgency and
difficulty in micturition. Contractions of ureter are also increased.
Uterus The action is
clinically insignificant, may slightly prolong
labour.
Bronchi Morphine releases
histamine which can cause bronchoconstriction.
This is of no consequence in normal individuals, but can be dangerous in
asthmatics.
6. ANS
Morphine causes mild
hyperglycaemia due to central sympathetic stimulation. It has weak
anticholinesterase action.
Pharmacokinetics
The oral absorption of morphine is unreliable because of high
and variable first pass metabolism; oral bioavailability is 1/6th to 1/4th of
parenterally administered drug. About 30% is bound to plasma proteins.
Distribution is wide; concentration in liver, spleen and kidney is higher than
that in plasma. Only a small fraction enters brain rather slowly. Morphine
freely crosses placenta and can affect the foetus more than the mother. It is
primarily metabolized in liver by glucuronide conjugation. Morphine6glucuronide
is an active metabolite (inherently more potent than morphine) which
accumulates during chronic dosing and contributes to analgesia, despite its
restricted passage across blood-brain barrier. Another metabolite morphine3glucuronide
has neuroexcitatory property. Plasma t½ of morphine averages 2–3 hours. Effect
of a parenteral dose lasts 4–6 hours. Elimination is almost complete in 24
hours and morphine is noncumulative. Small amounts may persist due to
enterohepatic circulation.
Adverse Effects
1. Side Effects Sedation, mental clouding, lethargy and other subjective effects which
may even be dysphoric in some subjects; vomiting is occasional in recumbent
patient; constipation is common. Respiratory depression, blurring of vision,
urinary retention (especially in elderly male) are other side effects. BP may
fall, especially in hypovolaemic patient and if he/she walks about.
2. Idiosyncrasy and Allergy Allergy is uncommon and anaphylactoid reaction is rare.
Urticaria, itch, swelling of lips are the manifestations. A local reaction at
injection site may occur due to histamine release.
3. Apnoea This may occur in the newborn when morphine is given to the mother during
labour. The bloodbrain barrier of foetus is undeveloped, morphine attains
higher concentration in foetal brain than in that of mother. Naloxone 10 μg/kg injected in the
umbilical cord is the treatment of choice.
4. Acute Morphine Poisoning It is accidental, suicidal or seen in drug abusers. In the nontolerant
adult, 50 mg of morphine i.m. produces serious toxicity. The human lethal dose
is estimated to be about 250 mg. Manifestations are extensions of the
pharmacological action.
Stupor or coma, flaccidity, shallow and occasional breathing,
cyanosis, pinpoint pupil, fall in BP and shock; convulsions may be seen in few,
pulmonary edema occurs at terminal stages, death is due to respiratory failure.
Treatment: consists of respiratory support (positive
pressure respiration also decreases pulmonary edema formation) and maintenance
of BP (i.v. fluids, vasoconstrictors). Gastric lavage should be done with pot.
permanganate to remove unabsorbed drug. Lavage is indicated even when morphine
has been injected; being a basic drug it is partitioned to the acid gastric
juice, ionizes there and does not diffuse back into blood.
Specific antidote: Naloxone 0.4–0.8 mg
i.v. repeated every 2–3 min till respiration picks up, is the preferred
specific antagonist because it does not have any agonistic action and does not per se depress respiration. It has a
short duration of action. Injection should be repeated every 1–4 hours later
on, according to the response. Nalorphine is no longer used.
5. Tolerance And Dependence High degree of tolerance can be developed to morphine and related
opioids if the drug is used repeatedly. It is partly pharmacokinetic (enhanced
rate of metabolism), but mainly pharmacodynamic (cellular tolerance). Tolerance
is exhibited to most actions, but not to constipating and miotic actions.
Addicts tolerate morphine in grams: lethal dose is markedly increased. Patients
in intense pain are relatively tolerant to depressant effects. Cross tolerance
among opioids is of high degree. Morphine tolerant subjects are partially cross
tolerant to other CNS depressants as well.
Morphine produces pronounced psychological and physical
dependence, its abuse liability is rated high. Recently the NMDA antagonists
and nitric oxide synthase inhibitors have been found to block morphine
tolerance and dependence in animals. Thus, analgesic action of morphine can be
dissociated from tolerance and dependence which contribute to its abuse.
Concern about abuse has been a major limitation in the use of morphine, but
appropriate medical use of morphine seldom progresses to dependence and abuse.
Morphine abuse is higher among medical and paramedical personnel. Earlier,
morphine addicts tended to be from the middle age group, but now younger
individuals are also opting for it. Opium eating has been prevalent among
natives in the orient.
Withdrawal of morphine is associated with marked drug seeking behaviour.
Physical manifestations are—lacrimation, sweating, yawning, anxiety, fear,
restlessness, gooseflesh, mydriasis, tremor, insomnia, abdominal colic,
diarrhoea, dehydration, rise in BP, palpitation and rapid weight loss. Delirium
and convulsions are not a characteristic feature (contrast barbiturates) and
are seen only occasionally. Cardiovascular collapse and fatality are rare if
supportive measures are instituted.
Opioid antagonists (naloxone, nalorphine) precipitate acute
withdrawal syndrome in the dependent subject. In the more severely dependent,
even 0.2 mg of naloxone can precipitate marked withdrawal.
Treatment: consists of
withdrawal of morphine and substitution with oral methadone (long-acting,
orally effective) followed by gradual withdrawal of methadone. However, relapse
rate among post-addicts is high. Long-term methadone maintenance and other
techniques using agonist-antagonistic drugs are also employed.
Precautions And Contraindications
Morphine is a drug of emergency, but due care has to be taken in
its use.
· Infants and the elderly are more susceptible
to the respiratory depressant action of morphine.
· It is dangerous in patients with respiratory
insufficiency (emphysema, pulmonary fibrosis, cor pulmonale), sudden deaths
have occurred.
·
Bronchial asthma: Morphine can precipitate an
attack by its histamine releasing action.
·
Head injury: morphine is contraindicated in
patients with head injury. Reasons are—
(a) By retaining CO2, it increases intracranial
tension which will add to that caused by head injury itself.
(b) Even therapeutic doses can cause marked respiratory
depression in these patients.
(c) Vomiting, miosis and altered mentation produced by morphine
interfere with assessment of progress in head injury cases.
·
Hypotensive states and hypovolaemia exaggerate
fall in BP due to morphine.
· Undiagnosed acute abdominal pain: morphine can
aggravate certain conditions, e.g. diverticulitis, biliary colic, pancreatitis.
Inflamed appendix may rupture. Morphine can be given after the diagnosis is
established. Pentazocine, buprenorphine are less likely to aggravate biliary
spasm.
·
Elderly male: chances of urinary retention are
high.
·
Hypothyroidism, liver and kidney disease
patients are more sensitive to morphine.
·
Unstable personalities: are liable to continue
with its use and become addicted.
Interactions
Phenothiazines, tricyclic antidepressants, MAO inhibitors,
amphetamine and neostigmine potentiate morphine and other opioids, either by
retarding its metabolism or by a pharmacodynamic interaction at the level of
central neurotransmitters.
Morphine retards absorption of many orally administered drugs by
delaying gastric emptying.
Dose: 10–50 mg oral, 10–15 mg i.m. or s.c. or 2–6 mg i.v.; 2–3 mg epidural/intrathecal; children
0.1–0.2 mg/kg.
MORPHINE SULPHATE 10 mg/ml inj; MORCONTIN 10, 30, 60, 100 mg
continuous release tabs; 30–100 mg BD; RILIMORF 10, 20 mg tabs, 60 mg SR tab.
CLASSIFICATION OF OPIOIDS
1. Natural
opium alkaloids: Morphine, Codeine
2. Semisynthetic
opiates: Diacetylmorphine (Heroin), Pholcodeine.
Many others like—Hydromorphone, Oxymorphone, Hydrocodone,
Oxycodone, are not used in India.
3. Synthetic opioids: Pethidine (Meperidine), Fentanyl, Methadone, Dextropropoxyphene,
Tramadol.
Many others like—Levorphanol, Dextromoramide, Dipipanone,
Alfentanil, Sufentanil, Remifentanil are not available in India.
Codeine
It is methylmorphine,
occurs naturally in opium,
and is partly converted in the body to morphine. It is less potent than
morphine (1/10th as analgesic), also less efficacious; is a partial agonist at μ opioid receptor with
a low ceiling effect. The degree of analgesia is comparable to aspirin (60 mg
codeine ~ 600 mg aspirin); can relieve mild to moderate pain only.
However, it is more selective cough suppressant (only 1/3rd as
potent as morphine); subanalgesic doses (10–30 mg) suppress cough (see p. 214). Codeine has very low affinity
for opioid receptors. The analgesic action has been ascribed to morphine
generated by its demethylation by CYP2D6; codeine fails to produce analgesia in
subjects with polymorphic CYP2D6. However, receptors involved in antitussive
action appear to be distinct, because they bind codeine as well as morphine.
Codeine has good activity by the oral route (oral: parenteral
ratio 1:2). A single oral dose acts for 4–6 hours. Constipation is a prominent
side effect when it is used as analgesic. Codeine has been used to control
diarrhoea (see Ch. No. 48). Other
side effects are milder. The abuse liability is low. Though codeine phosphate
is water soluble and can be injected, parenteral preparation is not available.
Pholcodeine
It has codeine like properties and has been used mainly as antitussive; claimed to be
less constipating.
Heroin
(Diamorphine, Diacetylmorphine)
It is about 3 times more
potent than morphine; more lipid soluble: enters brain more rapidly but
duration of action is similar. It is considered to be more euphorient
(especially on i.v. injection) and highly addicting. Because of its high
potency, it has been favoured in illicit drug trafficking. The sedative, emetic
and hypotensive actions are said to be less prominent. However, it has no outstanding
therapeutic advantage over morphine and has been banned in most countries
except U.K.
Pethidine (Meperidine)
Pethidine was synthesized as an atropine substitute in 1939, and
has some actions like it. Though chemically unrelated to morphine, it interacts
with opioid receptors and its actions are blocked by naloxone. Important
differences in comparison to morphine are:
·
Dose to dose 1/10th in analgesic potency;
however, analgesic efficacy approaches near to morphine and is greater than
codeine.
·
After i.m. injection, the onset of action is
more rapid but duration is shorter (2–3 hours).
·
It does not effectively suppress cough.
·
Spasmodic action on smooth muscles is less
marked—miosis, constipation and urinary retention are less prominent.
Pethidine is believed to induce less biliary spasm than
morphine; traditionally preferred in cholecystitis/biliary colic. However,
there is no objective evidence to support this belief. One study* in patients
undergoing cholecystectomy found pethidine to raise common bile duct pressure
14% more than equianalgesic dose of morphine.
· It is equally sedative and euphoriant, has
similar abuse potential. The degree of respiratory depression seen at
equianalgesic doses is equivalent to morphine.
·
Tachycardia (due to antimuscarinic action)
occurs instead of bradycardia.
·
It causes less histamine release and is safer
in asthmatics.
·
It has local anaesthetic action: corneal anaesthesia
is seen after systemic doses.
·
It is well absorbed, oral: parenteral activity
ratio is high (1/3 to 1/2). Pethidine is nearly completely metabolized in
liver. The plasma t½ of pethidine is 2–3 hours. Acidification of urine increases
excretion of unchanged pethidine.
Side Effects
These are similar to morphine except those mentioned
above. Some atropinic effects (dry mouth, blurred vision, tachycardia) may be
noted in addition.
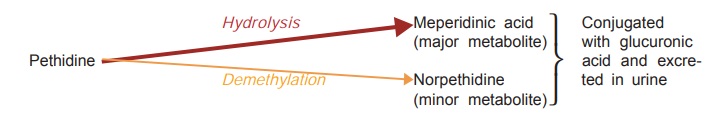
Overdose of pethidine
produces many excitatory effects—tremors, mydriasis, hyperreflexia, delirium,
myoclonus and convulsions. This is due to accumulation of norpethidine which has excitant effects. Renal failure patients
given repeated doses of pethidine may also experience similar effects.
Nonselective MAO inhibitors interfere with hydrolysis but not
with demethylation of pethidine—norpethidine is produced in excess and
excitement occurs.
Tolerance and physical dependence develop slowly with pethidine.
Probably due to its shorter duration of action, body functions get time to
recover. For the same reason withdrawal syndrome develops more rapidly.
Autonomic![]() disturbances are less marked during pethidine
withdrawal, than after morphine withdrawal.
disturbances are less marked during pethidine
withdrawal, than after morphine withdrawal.
Use
Pethidine is primarily
used as an analgesic (substitute of
morphine) and in preanaesthetic medication, but not for cough or diarrhoea. It
has also been used to control shivering during recovery from anaesthesia or that
attending i.v. infusions. Potential adverse effects due to accumulation of norpethidine
limit its utility in patients who require repeated dosing. It is the preferred
opioid analgesic during labour—at equianalgesic doses neonatal respiratory
depression is less marked, but still significant.
Dose: 50–100 mg i.m., s.c.
(may cause irritation, local fibrosis
on repeated injection), occasionally given orally or i.v.
PETHIDINE HCL 100 mg/2
ml inj; 50, 100 mg tab.
Fentanyl
A pethidine congener,
80–100 times more potent than
morphine, both in analgesia and respiratory depression. In analgesic doses it
produces few cardiovascular effects; has little propensity to release
histamine. Because of high lipid solubility, it enters brain rapidly and
produces peak analgesia in 5 min after i.v. injection. The duration of action is
short: starts wearing off after 30–40 min due to redistribution, while
elimination t½ is ~4 hr. In the injectable form it is almost exclusively used
in anaesthesia (see p. 376).
Transdermal fentanyl has become available for use in cancer or other types of
chronic pain for patients requiring opioid analgesia.
DUROGESIC transdermal
patch delivering 25 μg/hr, 50 μg/hr or 75 μg per hour; the patch
is changed every 2–3 days.
Methadone
A synthetic opioid,
chemically dissimilar but
pharmacologically very similar to morphine—has analgesic, respiratory depressant,
emetic, antitussive, constipating and biliary actions similar to morphine.
The most important
feature of methadone is high oral: parenteral activity ratio (1 : 2) and its
firm binding to tissue proteins. In single doses it is only slightly more potent
than morphine and has comparable duration of action (4–6 hours on i.m.
injection), but it cumulates in tissues on repeated administration—duration of
action is progressively lengthened due to gradual release from these sites;
plasma t½ on chronic use is 24– 36 hours. Plasma protein binding is 90% and it is
metabolized in liver, primarily by demethylation and cyclization—metabolites
are excreted in urine. Rifampin and phenytoin can cause withdrawal symptoms to
appear in methadone dependent subjects by inducing its metabolism.
Because of slow and
persistent nature of action, sedative and subjective effects are less intense.
It is probably incapable of giving a ‘kick’. The abuse potential is rated lower
than morphine. Tolerance develops more slowly, probably due to progressive
filling of tissue stores. Withdrawal syndrome is of gradual onset, taking 1–2
days after discontinuation, is prolonged and less severe.
Methadone has been
used primarily as substitution therapy of opioid dependence: 1 mg of oral
methadone can be substituted for 4 mg of morphine, 2 mg of heroin and 20 mg of
pethidine. Another technique is methadone
maintenance therapy in opioid addicts—sufficient dose of methadone is given
orally to produce high degree of tolerance so that pleasurable effects of i.v.
doses of morphine or heroin are not perceived and the subject gives up the
habit.
It can also be used as
an analgesic for the same conditions as morphine; dose 2.5–10 mg oral or i.m.
but not s.c. It is occasionally employed as antitussive.
PHYSEPTONE 10 mg inj,
2 mg/5 ml linctus.
Dextropropoxyphene
It is chemically related to methadone but is quite similar in
analgesic action and in side effects to codeine, except that it is a poor
antitussive and probably less constipating. It is nearly ½ as potent as codeine
and has a lower oral: parenteral activity ratio. It is metabolized in liver; t½
is variable (4–12 hours). Delirium and convulsions have occurred in overdose.
The demethylated metabolite of propoxyphene is cardiotoxic. The abuse liability
is similar to or lower than codeine.
Dextropropoxyphene (60–120 mg) is used as a mild oral analgesic.
It is marketed only in combination with paracetamol ± other drugs; but the
contribution of dextropropoxyphene to the analgesic effect of the combination
is questionable. The cardiac toxicity of its demethylated metabolite and
seizures are dangerous in overdose; only partly antagonized by naloxone.
Because of reported fatalities and no clear advantage of the combinations over
paracetamol alone, such preparations have been withdrawn in the UK, but are quite
popular in India, USA, etc, probably due to the perceived addictive potential
of codeine.
PARVODEX 60 mg cap: PARVON, PROXYVON, WALAGESIC:
dextropropoxyphene 65 mg + paracetamol 400 mg cap; WYGESIC, SUDHINOL 65 mg + paracetamol 650
mg cap.
Tramadol
This centrally acting analgesic relieves pain by opioid as well as additional
mechanisms. Its affinity for μ opioid receptor is low, while that for κ and δ is very low. Unlike
other opioids, it inhibits reuptake of NA and 5HT, and thus activates monoaminergic
spinal inhibition of pain. Its analgesic action is only partially reversed by
the opioid antagonist naloxone.
Injected i.v. 100 mg tramadol is equianalgesic to 10 mg i.m.
morphine; oral bioavailability is good (oral: parenteral dose ratio is 1.4).
The t½ is 5 hours and effects last for 4–6 hrs. Tramadol causes less
respiratory depression, sedation, constipation, urinary retention and rise in
intra-biliary pressure than morphine. It is well tolerated; side effects are
dizziness, nausea, sleepiness, dry mouth, sweating and lowering of seizure
threshold. Haemodynamic effects are minimal.
Tramadol is indicated for mild to moderate short-lasting pain
due to diagnostic procedures, injury, surgery, etc, as well as for chronic pain
including cancer pain, but is not effective in severe pain. Little tendency to
dose escalation is seen and abuse potential is low.
Dose: 50–100 mg
oral/i.m./slow i.v. infusion (children 1–2
mg/kg) 4–6 hourly.
CONTRAMAL, DOMADOL, TRAMAZAC 50 mg cap, 100 mg SR tab; 50 mg/ml inj
in 1 and 2 ml amps.
Uses (Of Morphine and its congeners).
As
analgesic
Opioid analgesics are
indicated in severe pain of any type. However, they only provide symptomatic
relief without affecting the cause. Pain may be valuable for diagnosis: should
not be relieved by analgesic unless proper assessment of the patient has been
done. Indiscriminate use of opioids can be hazardous. On the other hand, inadequate
dose or reluctance to use these drugs in a patient in distress is equally
deplorable.
Morphine or one of its parenteral congeners is indicated
especially in traumatic, visceral, ischaemic (myocardial infarction),
postoperative, burn, cancer pain and the like. It should be given promptly in
myocardial infarction to allay apprehension and reflex sympathetic stimulation.
Opioids, especially pethidine, have been extensively used for obstetric
analgesia, but one must be prepared to deal with the foetal and maternal
complications.
Adequate use of morphine (even i.v.) is indicated in an
emergency. It may prevent neurogenic shock and other autonomic effects of
excruciating pain. Opioids should not be restricted in case of pain of terminal
illness (cancer pain), but for other chronic conditions, due consideration must
be given to their addicting liabilities. Neuropathic pain responds less
predictably to opioid analgesics.
Epidural (2–3 mg) or
intrathecal (0.2 mg) injection of morphine produces segmental analgesia lasting
~12 hour without affecting other sensory, motor or autonomic modalities. It is
being used for surgical analgesia in abdominal, lower limb and pelvic
operations as well as for labour, postoperative, cancer and other intractable
pain. Respiratory depression occurs after a delay due to ascent of the opioid
through the subarachnoid space to the respiratory centre. Use of fentanyl in
place of morphine produces faster analgesia and reduces the risk of respiratory
depression because of greater uptake by nerves at the site of injection.
Patient controlled
analgesia (PCA) is an attractive technique of postoperative pain control in
which the patient himself regulates the rate of i.v. fentanyl infusion
according to intensity of pain felt.
Transdermal fentanyl
is a suitable option for chronic cancer and other terminal illness pain. The
patch produces analgesia after ~12 hr, but then blood levels of fentanyl and
intensity of analgesia remain fairly uniform if the patch is changed every 2–3
days.
For milder pain, e.g.
toothache, headache, neuralgias, etc., aspirin-like analgesics are preferred.
When they are not effective—codeine/ dextropropoxyphene may be used orally,
either alone or in combination with aspirin-like drug. The combination enhances
the ceiling analgesia. For majority of painful conditions, especially more
severe and longer-lasting pain, a NSAID should be combined with the opioid;
helps to enance analgesia while keeping the opioid dose low.
Preanaesthetic
Medication
Morphine and pethidine are used in few selected patients.
Balanced Anaesthesia
And Surgical Analgesia
Fentanyl, morphine,
pethidine, alfentanil or sufentanil are an
important component of anaesthetic techniques.
Relief Of
Anxiety And Apprehension
Especially in myocardial infarction, internal bleeding
(haematemesis, threatened abortion, etc.) morphine or pethidine have been
employed. However, they should not be used as anxiolytics or to induce sleep.
Acute Left
Ventricular Failure (Cardiac Asthma)
Morphine (i.v.)
affords dramatic relief by—
Reducing preload on
heart due to vasodilatation and peripheral pooling of blood.
Tending to shift blood
from pulmonary to systemic circuit; relieves pulmonary congestion and edema.
Allays air hunger by
depressing respiratory centre.
Cuts down sympathetic stimulation by calming the patient,
reduces cardiac work.
It is also indicated to relieve pulmonary edema due to
infarction of lung and other causes, but not due to irritant gases. It is contraindicated
in bronchial asthma.
Cough
Codeine or its
substitutes are widely used for suppressing
dry, irritating cough (see Ch. No.
16).
Diarrhoea
The constipating action of codeine has been used to check
diarrhoea and to increase the consistency of stools in colostomy. Synthetic
opioids exclusively used as antidiarrhoeals are diphenoxylate and loperamide.
The risk and benefits of their use are detailed in Ch. No. 48.
Related Topics
