Adrenergic Transmission
| Home | | Pharmacology |Chapter: Essential pharmacology : Adrenergic System and Drugs
Adrenergic (more precisely ‘Noradrenergic’) transmission is restricted to the sympathetic division of the ANS. There are three closely related endogenous catecholamines (CAs).
ADRENERGIC TRANSMISSION
Adrenergic (more precisely ‘Noradrenergic’) transmission is restricted to the sympathetic division of the ANS. There are three closely related endogenous catecholamines (CAs).
Noradrenaline (NA) It acts as transmitter at postganglionic sympathetic sites (except sweat glands, hair follicles and some vasodilator fibres) and in certain areas of brain.
Adrenaline (Adr) It is secreted by adrenal medulla and may have a transmitter role in the brain.
Dopamine (DA) It is a major transmitter in basal ganglia, limbic system, CTZ, anterior pituitary, etc. and in a limited manner in the periphery.
Synthesis of CAs
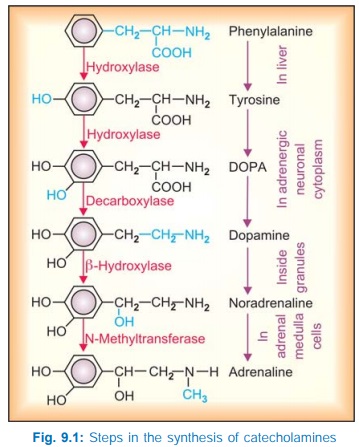
Catecholamines are synthesized from the amino acid phenylalanine as depicted in Fig. 9.1. Tyrosine hydroxylase is the rate limiting enzyme and its inhibition by αmethylptyrosine results in depletion of CAs; this can be used in pheochromocytoma before surgery and in inoperable cases. All enzymes of CA synthesis are rather nonspecific and can act on closely related substrates, e.g. dopa decarboxylase can from 5HT from 5hydroxytryptophan and α methyl DA from α methyl dopa. Synthesis of NA occurs in all adrenergic neurones, while that of Adr occurs only in the adrenal medullary cells. It probably requires high concentration of glucocorticoids reaching through intraadrenal portal circulation for induction of the methylating enzyme.
Storage of CAs
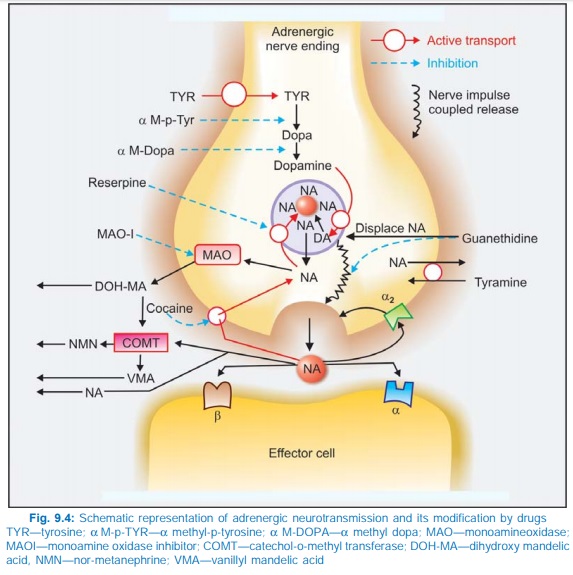
NA is stored in synaptic vesicles or ‘granules’ within the adrenergic nerve terminal (Fig. 9.4). The vesicular membrane actively takes up DA from the cytoplasm and the final step of synthesis of NA takes place inside the vesicle which contains dopamine βhydroxylase. NA is then stored as a complex with ATP (in a ratio of 4 : 1) which is adsorbed on a protein chromogranin. In the adrenal medulla the NA thus formed within the chromaffin granules diffuses out into the cytoplasm, is methylated and Adr so formed is again taken up by a separate set of granules. The cytoplasmic pool of CAs is kept low by the enzyme monoamine oxidase (MAO) present on the outer surface of mitochondria.
Release of CAs
The nerve impulse coupled release of CA takes place by exocytosis and all the vesicular contents (NA or Adr, ATP, dopamine β hydroxylase, chromogranin) are poured out. In case of vesicles which in addition contain peptides like enkephalin or neuropeptide Y (NPY), these cotransmitters are simultaneously released. The release is modulated by presynaptic receptors, of which α2 inhibitory control is dominant.
The autoreceptors of other cotransmitters (Y2 of NPY and P1 of ATP) also inhibit transmitter release. In addition, numerous heteroreceptors are expressed on the adrenergic neurone which either inhibit (dopaminergic, serotonergic, muscarinic and PGE2) or enhance (β2 adrenergic, angiotensin AT1 and nicotinic) NA release.
Indirectly acting sympathomimetic amines (tyramine, etc.) also induce release of NA, but they do so by displacing NA from the nerve ending binding sites and by exchange diffusion utilizing norepinephrine transporter (NET) the carrier of uptake1 (see below). This process is not exocytotic and does not require Ca2+.
Uptake of CAs
There is a very efficient mechanism by which NA released from the nerve terminal is recaptured. This occurs in 2 steps—
Axonal uptake An active amine pump (NET) is present at the neuronal membrane which transports NA by a Na+ coupled mechanism. It takes up NA at a higher rate than Adr and had been labelled uptake1. The indirectly acting sympathomimetic amines like tyramine, but not isoprenaline, also utilize this pump for entering the neurone. This uptake is the most important mechanism for terminating the postjunctional action of NA. This pump is inhibited by cocaine, desipramine and few other drugs.
Vesicular uptake The membrane of intracellular vesicles has another amine pump the ‘vesicular monoamine transporter’ (VMAT2), which transports CA from the cytoplasm to within the storage vesicle. The VMAT2 transports monoamines by exchanging with H+ ions. The vesicular NA is constantly leaking out into the axoplasm and is recaptured by this mechanism. This carrier also takes up DA formed in the axoplasm for further synthesis to NA. Thus, it is very important in maintaining the NA content of the neurone. This uptake is inhibited by reserpine, resulting in depletion of CAs.
Extraneuronal uptake of CAs (uptake2) is carried out by extraneuronal amine transporter (ENT or OCT3) and other organic cation transporters OCT1 and OCT2 into cells of other tissues. In contrast to NET this uptake transports Adr at a higher rate than NA, is not Na+ dependent and is not inhibited by cocaine, but inhibited by corticosterone. It is not of physiological or pharmacological importance.
Metabolism of CAs
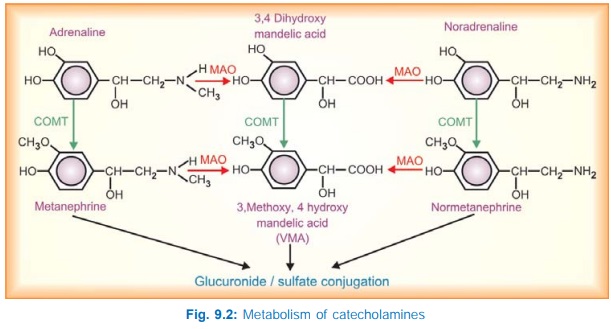
The pathways of metabolism of CAs are depicted in Fig. 9.2. Part of the NA leaking out from granules into cytoplasm as well as that taken up by axonal transport is first attacked by MAO, while that which diffuses into circulation is first acted upon by catecholomethyl transferase (COMT) in liver and other tissues. In both cases, the alternative enzyme can subsequently act to produce vanillylmandelic acid (VMA). The major metabolites excreted in urine are VMA and 3methoxy4hydroxy phenylethylene glycol (a reduced product) along with some metanephrine, normetanephrine and 3,4 dihydroxy mandelic acid. These metabolites are mostly conjugated with glucuronic acid or sulfate before excretion in urine. Only 25–50 μg of NA and 2–5 μg of Adr are excreted in the free form in 24 hours. However, metabolism does not play an important role in terminating the action of neuronally released CAs.
Adrenergic receptors
Adrenergic receptors are membrane bound Gprotein coupled receptors which function primarily by increasing or decreasing the intracellular production of second messengers cAMP or IP3/DAG. In some cases the activated Gprotein itself operates K+ or Ca2+ channels, or increases prostaglandin production.
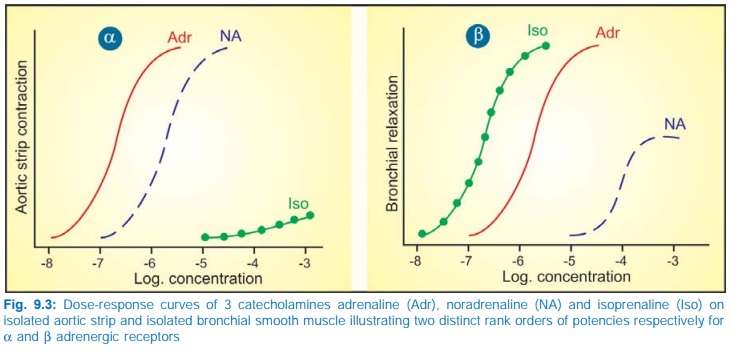
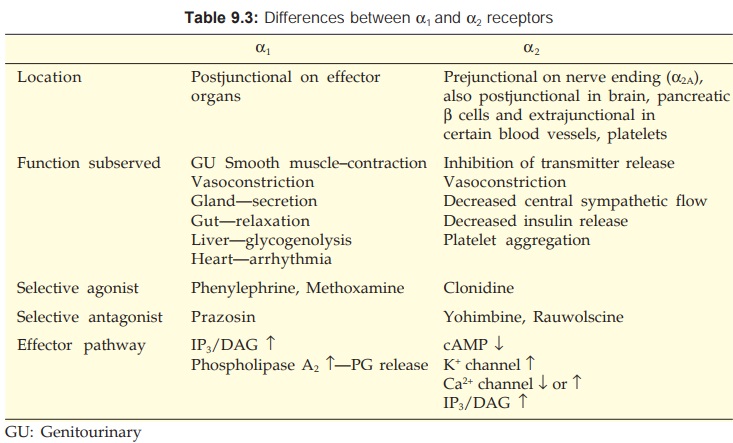
Ahlquist (1948), on the basis of two distinct rank order of potencies of adrenergic agonists (Fig. 9.3), classified adrenergic receptors into two types α and β. This classification was confirmed later by the discovery of selective α and β adrenergic antagonists. Important features of α and β receptors are given in Table 9.1.
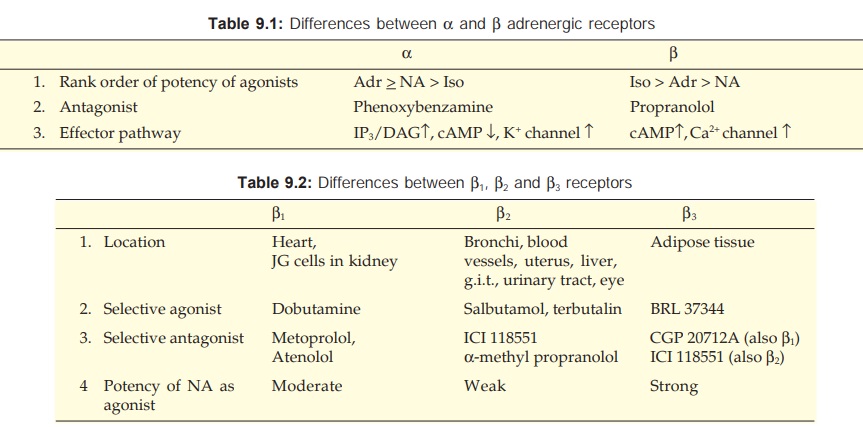
On the basis of relative organ specificity of selective agonists and antagonists the β receptors were further subdivided into β1 and β2 subtypes. Later, β3 (atypical β) receptors were described which have very low affinity for the standard blockers. These are located on adipocytes, mediate lipolysis and induce thermogenesis. Selective β3 agonists are being developed as potential antiobesity drugs.
In the mid 1970s the α receptors were demonstrated to be present prejunctionally as well. To differentiate these release inhibitory prejunctional α receptors, a subdivision into α1 and α2 was suggested. However, the present classification into α1 and α2 is based on pharmacological criteria (selectivity of agonists and antagonists) and not on anatomical location. Molecular cloning has further identified 3 subtypes of α1 (α1A, α1B, α1D) and 3 subtypes of α2 (α2A, α2B, α2C) receptors.
Though tissue distribution of subtypes of α1 and α2 receptors has been mapped, it is not very clear cut. Sufficiently subtype selective agonists or antagonists have also not yet been developed to pharmacologically exploit the molecular heterogeneity of α1 and α2 receptors.
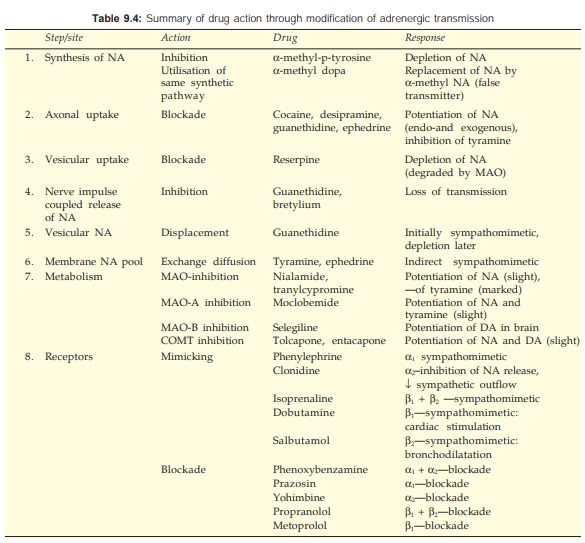
The adrenergic neuronal mechanisms and action of drugs which modify them are depicted in Fig. 9.4. A summary of drugs acting through adrenergic neuronal mechanisms is presented in Table 9.4
Related Topics
