Adverse Events in Clinical Trials
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Non-Clinical Safety Evaluation and Adverse Events in Phase I Trials
The safety monitoring that is designed into clinical trial protocols will include certain routine procedures that are used to monitor health and well-being of the study subjects, including the vital signs, routine haematology and clinical chemistry.
ADVERSE EVENTS IN CLINICAL
TRIALS
The
safety monitoring that is designed into clinical trial protocols will include
certain routine procedures that are used to monitor health and well-being of
the study subjects, including the vital signs, routine haematology and clinical
chemistry. Additional tests and monitoring methods may also be included in the
protocol to allow early identification of adverse events that may be attributed
to toxicities revealed by the non-clinical studies, for example additional
ECG/Holter monitoring and additional assays for specific biomarkers in blood or
urine.
In
first-into-human and other early phase clinical studies, it may be possible to
identify some likely adverse events from the non-clinical results and there are
always likely to be adverse events such as colds and headaches that may or may
not be related to the investigational product. It remains true, however, that
the profile of events for a given investigational product remains uncertain
until the safety data have been gathered and analysed. As such, care must be
taken to consider all events carefully and to assess causality. In early trials
with few subjects, it would be easy to overlook rare or mild effects that may
prove to be important signals when larger, more mixed and less healthy
populations are exposed to the product.
A
review of all adverse events recorded in volun-teers during two separate
12-month periods (1993 and 1998) at the Clinical Trials Unit of Charles River
Laboratories Clinical Services International Ltd (formerly Inveresk Research)
has been conducted. All adverse events reported spontaneously, elicited by
staff questioning, or observed were collected. Two doctors performed the allocation
of each event to the trial medication independently with a third arbiter in
cases of disagreement. The doctors were blinded to the study medication and
allocated causality according to the known pre-clinical pharmacology and
toxicology of the drug and the timing of the adverse event.
Of
a total of 30 studies (32 drugs) available for review in 1998, 10 were single
ascending dose toler-ability studies and 5 were multiple-dose tolerability
studies. The remaining studies were pharmacokinetic studies. Several
therapeutic classes of drugs were represented. Drug-related adverse events were
those considered possibly, probably or definitely related to the medication.
Data were compared with those collected in 1993 involving a total of 23 studies
(18 drugs). Comparison of the numbers of studies and exposures is made in Table
7.1 and details of the adverse events are given in Tables 7.2–7.4.
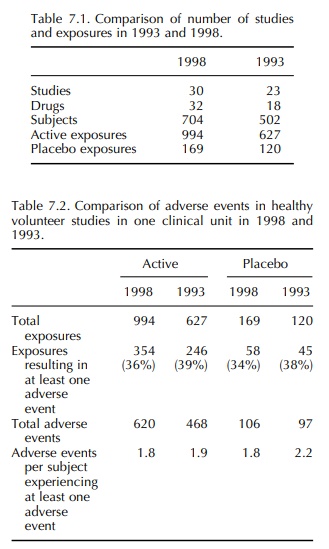
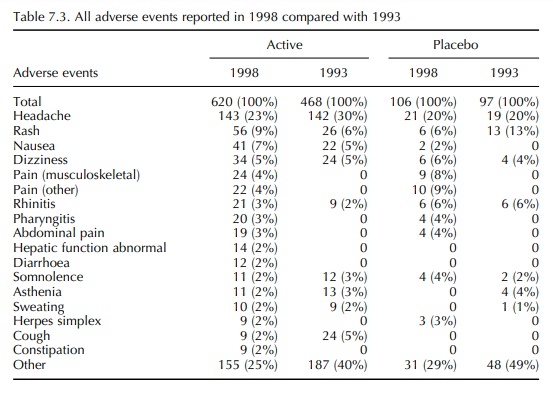
In the 1993 report, the frequency of adverse events reported in volunteer studies was much greater than that reported by Orme et al. (1989). However, the incidence reported in 1993 was confirmed by the 1998 data. It is also notable that, in 1993, there was a similar frequency of adverse events in volun-teers receiving an active drug and those receiving a placebo. This differs from the findings of Sibille et al. (1992), who reported a difference in the incidence of adverse events between active drug and placebo treat-ment, active being significantly higher. Once again, the 1993 results were confirmed in 1998. It is therefore concluded that the incidence of adverse event report-ing in healthy volunteer studies is 34%–39% with an almost identical incidence in placebo exposures as in active exposures. Most adverse events were mild and self-limiting, and in both 12-month periods the most common event in both active and placebo exposures was headache (19%–30%).
The
experience of Charles River Laboratories Phase I clinic over a 10-year period
from 1996 to 2005 indicates that the incidence of treatment-emergent Serious
Adverse Events in Phase I studies in healthy volunteers is low; approximately 1
– 2/1000 for volun-teers exposed to one or more doses of active or placebo drug
(See Table 7.5). The majority of these serious adverse events fall into the
category of a medically important event. This is similar to the incidence
reported in a survey by The Association of Independent Clinical Research
Contractors (AICRC) who reported an incidence of 1 in 509 (0.2%) from a total
of 128 in 65 205 subjects (AICRC members’ communication, 1999). Only one of the
serious adverse events reported by Charles River Laboratories over 10 years, a
low grade biochemical hepatitis of probable immunological pathogenesis in an
asymptomatic subject, was consid-ered to be probably drug related. This case
occurred in a late Phase I study; several thousand patients had received the
drug in clinical development, no similar cases had been reported and there was
no evidence of hepatoxicity in non-clinical studies. Two further SAEs were
considered possibly related to the IMP.
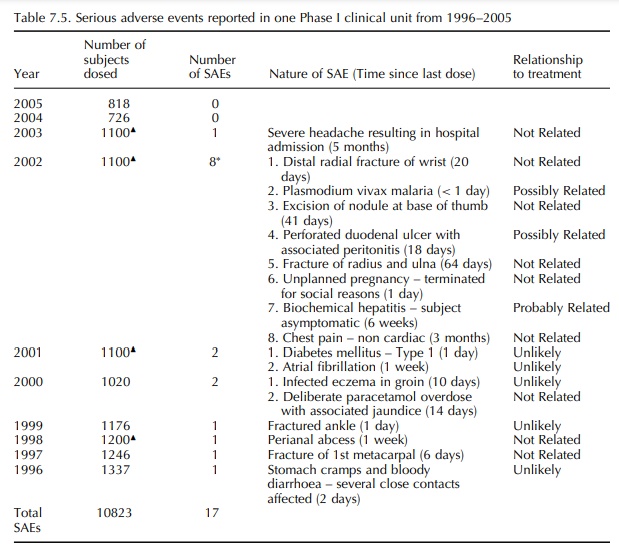
The
incidence of a single ‘probably related’ and two ‘possibly related’ serious
adverse events in 10 823 subjects (0.03%) indicates that current standards for
pre-clinical safety testing of new drugs are success-ful in ensuring that early
drug development studies in humans are safe and that the risks to individuals
subjects are relatively low.
The adverse event data gathered in Phase I and other early phase trials are essential to expanding the safety profile of the drug product and may be considered as an extension to the non-clinical data.
Critical
assessment of the possible relationship of an adverse event to the product is
fundamental to detecting signals of toxicity and to correctly assess-ing drug
safety prior to moving to larger scale trials. When reviewing the causality of
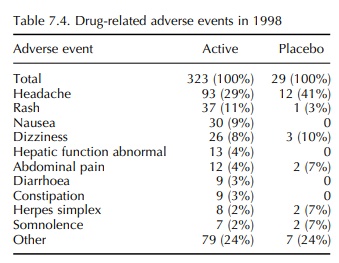
adverse events, it is notable that the 1998 data on adverse events that were
considered to be drug related (possibly, proba-bly or definitely) reveals that
certain adverse events, most notably headache, occurred at higher incidence in
subjects receiving placebo than in subjects receiv-ing active (Table 7.4). In
contrast, those events which can be measured in animals, for example
constipation, diarrhoea and abnormal liver function tests, feature only in the
active group. It may be concluded that these sorts of events are more reliable
indicators of drug effect and may differentiate active from placebo.
Attribution
of or, conversely, discounting relation-ship of an event to treatment should,
however, be done with caution. It is known that elevations in transaminase
concentrations can occur in healthy volunteers for several reasons, including
excess calo-rie intake, relative to normal, due to reduction in physical
activity (Kanamaru et al., 1989;
Purkins et al., 2004). Similar
findings have been noted at approximately
equal prevalence in both active- and placebo-treated subjects during periods of
residence of ≥ 7 days in this clinic (Wyld, 1991;
Unpublished internal study conducted at Charles River Laborato-ries, 2003). In
the 2003 study, data was collected from nine Phase I, randomised, double-blind,
placebo-controlled multiple-dose tolerance studies, in which, doses were
administered during at least 7 days of confinement. Over 300 subjects were
included. Clini-cally significant (CS) abnormalities were defined as > 1 5
times the upper limit of normal and were reported as adverse events. The incidence
of CS abnormal-ities for alanine aminotransferase (ALT) was 9% for active- and
8% for placebo-treated subjects. For aspartate aminotransferase (AST) the
incidences were 2% and 1% for active- and placebo-treated subjects,
respectively. These differences were not statistically significant (ALT p = 0 850; AST p = 0 652) when analysed using the
Chi-squared test.
The
liver function test data highlight the need to take all factors into account
when assessing the cause of an adverse event. Although increases in liver
function tests may be an artefact caused by changes to the diet, exercise and
environment of volunteers in clinical studies, the findings must be carefully
consid-ered before ruling out adaptive changes or toxicity. Only small numbers
of volunteers are used in Phase I clinical trials so it is important to review
the data gathered in all the clinical trials performed with a new
pharmaceutical, identifying consistencies across the studies. It is notable
that hepatic toxicity was, together with hypersensitivity/skin reactions, the
human toxi-city with the poorest correlation with animal studies in Olson’s
review (Olson, 2000). These were also the two toxicities that led most often to
termination of clinical development.
Further
study of mechanisms underlying such toxi-cities is required, especially in view
of the number of drugs that have been withdrawn or have had warnings added to
their labels due to hepatotoxicity. In the case of troglitazone (an
antidiabetic drug, voluntarily withdrawn from the market by the licence holder
in 2000), it was reported that 1.95% of patients treated with troglitazone in
clinical trials developed eleva-tions of aminotransferases that were greater
than three times the upper limit of normal. A similar finding was noted in 0.6%
of placebo-treated patients, so the increase seen in the active-treatment
groups seems quite modest (Lin, Chern and Chu, 2003). In the light of
subsequent incidences of serious idiosyncratic liver toxicity associated with
this product whilst on the market, it would seem that when there are
differences in the incidence of liver enzyme elevation between active- and
placebo-treated groups in clinical trials, this should be considered a signal
to examine the clinical and non-clinical data very closely before licensing.
This
example highlights the danger of missing early signals of toxicity that may, in
themselves, appear to be mild and of little or no significance but which may be
early signals of a toxicity which will be problem-atic in some individuals. It
is equally clear that the same minor events may well be of no toxicological or
clinical significance and should not hinder the clinical development of the
product in question. It is impor-tant, therefore, to consider the non-clinical
and early clinical safety data carefully as it becomes available and relate it
to the previous study data in order to identify any trends. If data from an
individual study are examined in isolation, it is easy to write off some mild
effects that may not have clear statistical signifi-cance or may not show a
marked dose relationship or related pathology. For example, a minor increase in
liver function enzymes in a rodent toxicology study, with no clear dose
relationship, may not cause any undue concern if no associated macroscopic or
microscopic changes in the liver are observed. If, however, simi-lar minor
effects were seen in a second species, it may not be sufficient to stop the
transition to humans but it would be appropriate to monitor the liver enzymes
carefully in early phase clinical studies. A similar prin-ciple applies as the
product progresses through clinical trials, increasing the safety database.
Minor increases in liver enzymes seen in Phase I trials in volunteers may be
related to changes in diet, exercise pattern and environment, as noted above,
but the possibility of a drug-induced change should not be ruled out,
especially if similar changes have been noted in the non-clinical studies.
Careful monitoring and surveillance of the data from subsequent studies will be
required to deter-mine the nature and ramifications of this type of effect. In
effect, clinical safety studies and, if the product progresses to market,
pharmacovigilance form a continuum with the non-clinical safety studies.
Related Topics
