Type a Adverse Drug Reactions
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Mechanisms of Adverse Drug Reactions
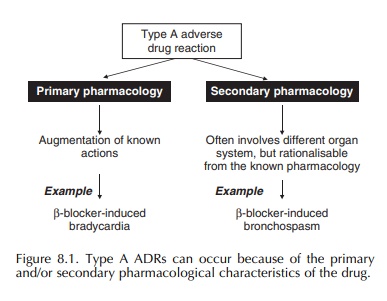
Pharmacological (type A) ADRs are the most common forms of drug toxicity.
TYPE A ADVERSE DRUG REACTIONS
Pharmacological
(type A) ADRs are the most common forms of drug toxicity (Pirmohamed et al., 1998). They can occur because of
the primary and secondary pharmacological characteristics of the drug (Figure
8.1). More emphasis is now placed on the secondary pharmacology of new drugs
during pre-clinical evaluation, to anticipate and thus avoid problems that
might arise once the drug is introduced into humans.
The experience with fialuridine, an experimental drug for hepatitis B, highlights the need for continued development of appropriate in vivo and, bridging, in vitro test systems for the prediction of secondary pharmacological adverse effects in humans. In June 1993, during phase II trials, 5 of 15 patients given fialuridine died, whereas two others required emer-gency liver transplants (McKenzie et al., 1995). The toxicity was delayed with patients presenting weeks to months after stopping fialuridine. The toxicity had not been observed in four animal species, and the only model seems to be the hepatitis B-infected wood-chuck. In vitro studies in cultured hepatoblasts have shown that the toxicity is because of the inhibition of DNA polymerase γ by fialuridine and its metabolites leading to mtDNA depletion and mitochondrial ultra-structural defects (Lewis et al., 1996).
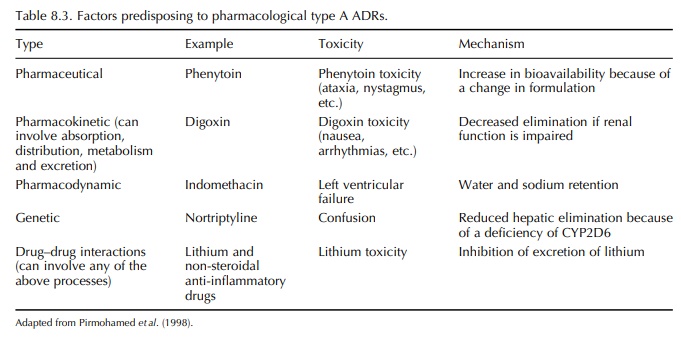
Factors
predisposing to pharmacological adverse reactions include dose, pharmaceutical
variation in drug formulation, pharmacokinetic or pharmaco-dynamic
abnormalities and drug–drug interactions (Pirmohamed et al., 1998) (Table 8.3). In essence, type A reactions occur when
the drug concentration in plasma or tissue exceeds the perceived therapeutic
window. Alternatively, the drug concentration may be within the normal range
defined for the population, but because of increased sensitivity of the target
in the individual, an adverse reaction results. There are many examples of drugs
(e.g. captopril) that had been introduced into clinical practice at a dose that
was subsequently shown to be associated with an unac-ceptable frequency of
ADRs, and for which a lower dose was found to be both safe and effective. In
general, however, the individual affected by a type A adverse reaction will
have impairment of clearance or increased sensitivity because of the normal
process of ageing, disease, concomitant drugs or genetic vari-ation or a
combination of these factors (Brodie and Feely, 1991).
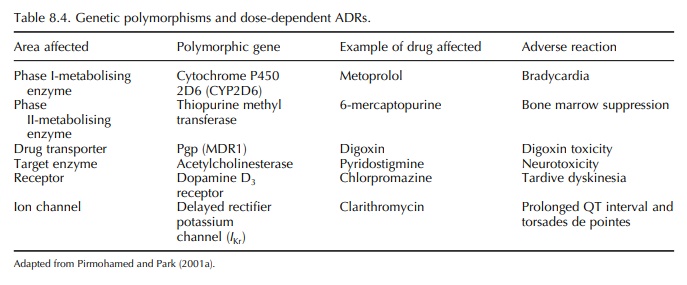
GENETIC POLYMORPHISMS AND TYPE A ADVERSE DRUG REACTIONS
A
gene can be defined as exhibiting genetic poly-morphisms if the variant allele
exists in the normal population at a frequency of at least 1%. Genetic
poly-morphisms are a source of variation to drug response in the human body. In
relation to type A ADRs, poly-morphisms in both pharmacokinetic and
pharmaco-dynamic parameters can act as predisposing factors (Table 8.4).
To date, most attention has focused on geneti-cally mediated deficiencies of the drug-metabolising enzymes (Park, 1986; Pirmohamed and Park, 1996). A drug metabolised by this pathway will show reduced elimination from the body with a consequent increase in half-life. This will lead to dose-dependent toxicity; a typical example is neutropenia with azathioprine in patients deficient in the enzyme thiopurine methyl-transferase (Lennard et al., 1982).
The
role of genetic variation in the metabolism of warfarin by CYP2C9 has attracted
a great deal of attention recently. Warfarin is the oral anticoagulant of
choice in the United Kingdom (Hart et al.,
1998). The number of patients attending anticoagulant clinics has doubled in
the last decade or so, largely because of its use in atrial fibrillation. The
major risk of warfarin treatment is haemorrhage with an incidence of 8–26 per
100 patient-years (Petty et al.,
1999); this is related to the intensity of anticoagulation. Minimi-sation of
the risk of bleeding depends on accurate clin-ical prediction of dosage
requirements during warfarin therapy. However, this is difficult because there
is 20-fold interindividual variability in the dose necessary to maintain the
international normalised ratio (INR) within a target range.
The
S-enantiomer of warfarin, which is predom-inantly responsible for the
anticoagulant effect, is metabolised by CYP2C9 (Rettie et al., 1992). Poly-morphisms in the CYP2C9 gene result in at least
two allelic variants, CYP2C9∗2 Arg144 → Cys and CYP2C9∗3 Ile359 → Leu (Furuya et al., 1995), both of which have been shown to decrease warfarin
clearance in vitro (Haining et al., 1996; Takahashi et al., 1998) and in vivo (Takahashi et al.,
1998). Clinically, these variants have been shown to be associated with a reduced
warfarin dose requirement, greater diffi-culty in initiating warfarin treatment
and an increased risk of bleeding (Aithal et
al., 1999). The strong and consistent relationship between CYP2C9 geno-type and dose requirement
has been confirmed in a systematic review. CYP2C9
genotype also seems to be important with respect to warfarin-related bleeding,
but the association is not as strong as that observed with dose (Sanderson,
Emery and Higgins, 2005). More recently, it has also been shown that
poly-morphisms in the gene-encoding vitamin K epoxide reductase complex 1 (VKORC1), the target for the action of
warfarin, also determine dose requirements (Rieder et al., 2005; Sconce et al.,
2005; Wadelius et al., 2005). Indeed,
the effect of VKORC1 seems to be quantitatively greater than that of CYP2C9. A limited subset of
environmental determinants (includ-ing age) and polymorphisms in the VKORC1 and CYP2C9 genes account for approximately 55% of the variance in warfarin dose requirements
(Rieder et al., 2005; Sconce et al., 2005; Wadelius et al., 2005). Sconce et al. (2005) have recently gone onto
develop a dosing table based on a regression equation combin-ing age, height
and CYP2C9 (∗2 and ∗3) and the VKORC1 (−1639G > A)
single-nucleotide polymor-phisms (SNPs). Whether such genotype-based dosing, in
the absence of other possible factors that might influence dose requirements,
including drug interac-tions, diet, underlying disease, e.g. thyroid disease,
and polymorphisms in other genes involved in the mode of action of warfarin,
will lead to an improve-ment in the dosing and safety of warfarin, requires
further study (Pirmohamed and Park, 2001a).
DRUG INTERACTIONS AND ADVERSE DRUG REACTIONS
Patients
on polytherapy are more likely to have type A reactions. The likelihood of
developing an adverse interaction increases with the number of drugs prescribed
(D’Arcy, 1986). To date, this has largely been a problem in the elderly where
polyphar-macy is prevalent (Williamson and Chopin, 1980) but is becoming
increasingly frequent in younger patients with chronic diseases such as AIDS,
where patients may be on 6–10 different drugs (Bayard, Berger and Jacobson,
1992). An Australian study showed that 4.4% of all ADRs resulting in hospital
admission were because of drug interactions (Stanton et al., 1994), whereas a study in the United Kingdom showed that one in six of all adverse
reactions caus-ing hospital admission were because of interactions (Pirmohamed et al., 2004).
Drug
interactions due to effects on metabolic path-ways may be because of either
enzyme induction or enzyme inhibition (Brodie and Feely, 1991). Enzyme
induction usually leads to increased metabolism of the drug and thus increases
drug clearance. This will lead to reduced drug efficacy rather than drug
toxicity (unless the adverse reaction is because of a metabo-lite rather than
the parent drug). Enzyme inhibition on the contrary is more likely to lead to
type A ADRs because the clearance of the affected drug is reduced; this is
particularly likely when the affected drug has a narrow therapeutic index
(Brodie and Feely, 1991). Indeed, enzyme inhibitory drug inter-actions have
resulted in regulatory action in many instances. An important example was the
interac-tion between the CYP3A4 inhibitors ketoconazole and erythromycin and
the non-sedating antihistamine terfenadine (Konig et al., 1992; Woosley et al.,
1993). This resulted in decreased conversion of terfenadine to its active
metabolite (now marketed as fexofenadine). Terfenadine has been shown to affect
the delayed rectifier potassium current (Chen, Gillis and Woosley, 1991), which
results in the prolongation of QT inter-val, torsades de pointes and sudden
death. A simi-lar interaction with cisapride and CYP3A4 inhibitors (Michalets
and Williams, 2000) has also resulted in regulatory action against cisapride.
Interestingly, such enzyme inhibitory interactions can also occur with foods
such as grapefruit juice and cranberry juice.
A
new mechanism of adverse interaction involves drug transporters in the
disposition of drugs. Many drug transport proteins are present on membranes,
some of which are responsible for drug influx and some are responsible for drug
efflux, whereas others can transport in both directions. Most of the focus to
date has been on P-glycoprotein (Pgp), which is encoded by the multi-drug
resistance 1 (MDR1) gene.
Overexpression of Pgp is one of the mech-anisms responsible for resistance of
tumours to chemotherapy (Germann, 1996). However, Pgp is also responsible for
the transport of many other drugs including digoxin. Digoxin does not undergo
any significant degree of metabolism but interacts with drugs such as
quinidine, verapamil and amiodarone, all of which can precipitate digoxin
toxicity. The mechanism of this interaction involves the inhibition of Pgp,
thereby reducing efflux of digoxin from the gut and kidney (Fromm et al., 1999). As knowledge of the
transporters and their drug substrates increases, it is likely that this will
be identified as the mechanism underlying many adverse drug interactions.
Related Topics
