An Example of a Drug That Causes Toxicity Through the Formation of a Chemically Reactive Intermediate
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Mechanisms of Adverse Drug Reactions
TYPE B OR IDIOSYNCRATIC ADVERSE DRUG REACTIONS : Paracetamol: An Example of a Drug That Causes Toxicity Through the Formation of a Chemically Reactive Intermediate
PARACETAMOL: AN EXAMPLE OF A
DRUG THAT CAUSES TOXICITY THROUGH THE FORMATION OF A CHEMICALLY REACTIVE
INTERMEDIATE
For
many drugs that undergo metabolism, CRM will be formed irrespective of the dose
of the drug (Pirmohamed, Madden and Park, 1996). When a drug is taken in
therapeutic dosage, any toxic metabo-lite formed will be detoxified by normal
enzymatic or non-enzymatic cellular defence mechanisms. An imbalance between
bioactivation and bioinactivation leading to toxicity may however be created by
taking a drug overdose. This will lead to the formation of large amounts of
CRM, overwhelm the cellular detoxication capacity and lead to cell damage. The
clearest exam-ple of this is paracetamol, which causes hepatotoxicity when
taken in overdosage, and still causes about 160 deaths per year in the United
Kingdom (Bray, 1993). According to the conventional definition of ADRs,
paracetamol hepatotoxicity should not be classified as an ADR, because the
hepatic injury occurs when the drug is used inappropriately. However, it is
impor-tant to note that the occurrence of liver damage with paracetamol and its
severity is a function not only of the dose but also of various host factors
(Pirmohamed, Kitteringham and Park). Indeed, paracetamol hepato-toxicity has
been reported with therapeutic drug use. For example, a recent study in 67 alcoholics
who had sustained liver injury after paracetamol ingestion showed that 40% had
taken less than 4 g/day (the maximum recommended therapeutic dose), whereas
another 20% had taken between 4 and 6 g/day (which is also regarded as a
non-toxic dose) (Zimmerman and Maddrey, 1995).
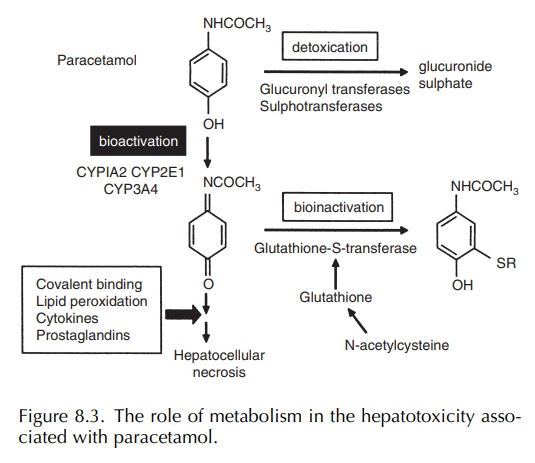
In
therapeutic dosage, paracetamol is largely metabolised by phase II processes
(glucuronidation and sulphation) to stable metabolites, but between 5% and 10%
also undergoes P450 metabolism to the toxic N -acetyl p-benzoquinoneimine
(NAPQI) metabolite (Nelson, 1990) (Figure 8.3). This is detox-ified by cellular
glutathione. In overdosage, satura-tion of the phase II metabolic pathways
results in a greater proportion of the drug undergoing bioactiva-tion. This
ultimately leads to the depletion of cellular glutathione and allows the toxic
metabolite to bind to hepatic proteins resulting in hepatocellular damage
(Nelson, 1990). The use of N -acetylcysteine in the treatment of paracetamol
overdosage illustrates the important point that elucidation of the mechanism of
drug toxicity can lead to the development of ratio-nal therapies that will
prevent the toxicity. Alcoholics show increased susceptibility to paracetamol
over-dosage because excess alcohol consumption results in the depletion of
glutathione (Lauterburg and Velez, 1988) and induction of the P450 isoform
CYP2E1 (Raucy et al., 1989). Recent
studies in knockout mice have shown that CYP2E1 is the primary isoform involved
in the bioactivation of paracetamol (Lee et
al., 1996).
Although
experiments with transgenic mice have shown that in the absence of phase I
oxidative pathways and therefore NAPQI formation, hepato-toxicity does not
occur, the precise pathway lead-ing to liver damage is still unclear (Gibson et al., 1996). Several mechanisms have
been proposed, including effects on plasma membrane Ca2+ pumps (Tsokos-Kuhn,
1989), which can lead to Ca2+-induced DNA damage (Ray et al., 1990), mito-chondrial damage
(Meyers et al., 1988) resulting in
glutathione depletion and oxidative stress (Jaeschke, 1990) and apoptosis (Ray et al., 1996). Recently, it has been
shown that Fas antisense oligonucleotide protects mice from paracetamol
toxicity, suggest-ing that the ultimate cytotoxic event involves more than
simply necrosis and that cells of the immune system may be recruited in the
inflammatory response (Zhang et al.,
2000). Interestingly, several studies have revealed that cells exposed to
chemical or oxidant stress will respond with an orchestrated and robust
transcriptional response aimed at detoxify-ing the offending chemical and
preventing or repair-ing cellular damage (Hayes et al., 1999; Moinova and Mulcahy, 1998, 1999). If unsuccessful,
then the culmination of this response, known as the antiox-idant response, is
to commit the cell to suicide through apoptosis. The target genes for the
antioxidant response encode a set of enzymes and other proteins that scavenge
free radicals, neutralise electrophiles or up-regulate the critical cellular
thiol, glutathione. Glutathione depletion caused by a range of chemicals leads
to the up-regulation of c-jun and c-fos mRNA and enhances activator protein-1
(AP-1) DNA bind-ing activity (Kitteringham et
al., 2000). This response was also accompanied by the induction of
-glutamyl cysteine synthetase (GCS). Another important mech-anism of cell
protection involves the nuclear translo-cation of redox-sensitive transcription
factors such as Nrf-2, which ‘sense’ chemical danger and orches-trate cell
defence. Importantly, it has been observed that nuclear translocation occurs at
non-toxic doses of paracetamol and at time points before overt toxi-city is
observed. However, with increasing doses of acetaminophen, there is progressive
dislocation of nuclear translocation, transcription, translation and protein
activity as the rate of drug bioactivation over-whelms cell defence through the
destruction of crit-ical proteins – at least 31 of these critical proteins have
been identified (Park et al., 2005).
Paradoxically,
studies performed with transgenic mice aimed at clarifying events subsequent to
NAPQI formation have only served to confound rather than to clarify. For
example, the deletion of compo-nents of the glutathione detoxication system
such as glutathione peroxidase (Mirochnitchenko et al., 1999) and glutathione transferase-pi (GST-pi) (Henderson et al., 2000) both afforded partial
protection against paracetamol
hepatotoxicity. The loss of a major hepatic form of GST, which represents over
3% of total soluble protein (Fountoulakis et
al., 2000), would have been expected to predispose the animals to
hepa-totoxicity through a reduction in the glutathione conju-gation of NAPQI
(Coles et al., 1988). This suggests
that GST-pi may be involved in a novel mechanism that determines susceptibility
to paracetamol hepato-toxicity. Indeed, a recent study has shown that GST-pi
may have a role in cell signaling; it has been shown to be an efficient
inhibitor of Jun kinase (also known as stress-activated kinase), the enzyme
that activates c-jun and several other transcription factors (Adler et al., 1999). Future studies using
other transgenic mouse models will be useful in determining the exact path-way
by which paracetamol causes liver damage and may therefore provide novel
therapeutic strategies by which to reverse liver damage in patients who present
late after paracetamol overdosage.
Related Topics
