Hammett Equation
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Mechanisms of Organic Reactions
The Hammett equation is a very useful tool for monitoring the extent of charge development in the activated complex.
HAMMETT EQUATION
The
Hammett equation is a very useful tool for monitoring the extent of charge
development in the activated complex. If a substituent is attached to the meta
or para position of a benzene ring, it will change the ability of the aromatic
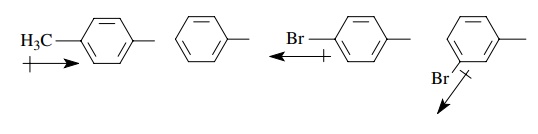
ring to donate or withdraw electrons relative to benzene itself. Thus a p-methylphenyl group should be electron
donating relative to phenyl while a p-bromophenyl
group should be electron withdrawing relative to phenyl. Furthermore, placement
of the bromo group in the meta position places it closer to the point of
attachment of the phenyl ring so the electron-withdrawing ability of the m-bromophenyl group is greater than that
of the p-bromophenyl group.
Substituents are normally attached only to the meta or para positions of the
phenyl so that they do not sterically interfere with the site of attachment of
the phenyl group.
By
the use of a model reaction (ionization of benzoic acids), the ability of a
substituent to modify the electron-donating or electron-withdrawing ability of
the phenyl group and thus influence that reaction can be defined quantitatively
by the Hammett equation,
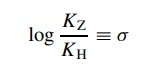
where
KZ is the acidity constant
for a benzoic acid with some substituent– Z attached and KH is the acidity constant for benzoic acid itself.
The result is a substituent constant (σ ) which is a numerical description of the electronic effect of a substituent relative to a hydrogen atom on the model reaction. Stated a different way, a substituent constant σ is a quantitative way to describe the electron-donating or electron-withdrawing properties of a substituent when it is attached to a benzene ring.
Different σ
values are obtained for a given substituent if it is in the meta or the para
position so σ values are positionally
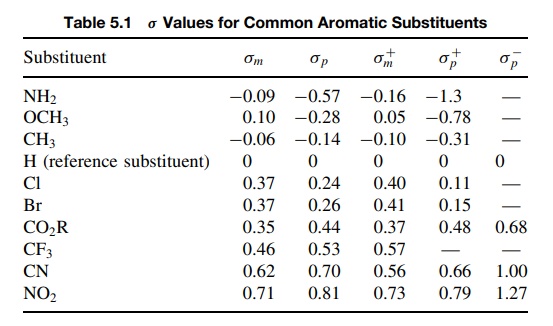
dependent. Table 5.1 lists some common σ
values.
The
model reaction used to evaluate σ
constants is an ionization equilibrium in which a negative charge is produced
upon going from reactants to products. Electron-withdrawing groups have
positive σ values because they
increase the ionization relative to hydrogen, KZ/KH
> 1, and log(KZ/K H) > 0. Electron-donating substituents
have negative σ values because they
decrease ionization relative to hydrogen, KZ/KH < 1, and log(KZ/KH) < 0. Hydrogen itself is treated as a substituent with σ = 0 because KZ =
KH and thus KZ/KH =
1 and log(KZ/K H) = 0.
Furthermore
the absolute magnitude of the σ value
provides a quantitative measure of the relative electron-donating or electron-withdrawing
ability. Thus a m-CF3 group (σ = 0.46) is a
stronger electron-withdrawing group than
m-Cl (σ = 0.37) but is a less strong
electron-withdrawing group than m-CN
(σ = 0.62). On the other hand, a p-methyl
group (σ = −0.14) is a weaker electron donor than p-OCH3 (σ = −0.28) but a better electron donor than p-Si(CH3)3 (σ = −0.07)
These
substituent constants can be used with rate data to evaluate the type and
extent of charge development in the activated complex of the rate-determining
step for a wide variety of chemical reactions. The rates of reaction for a
particular transformation are measured using a series of compounds which differ
only by the phenyl substituents present; for example,
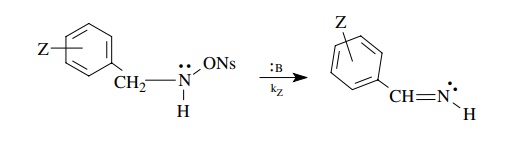
Then
the differences in rate caused by the electronic effect of the substituent are
correlated by the Hammett equation log(kz/ kH) = ρσz, where kz is the rate constant
obtained for a compound with a particular meta or para substituent, kH is the rate constant for the unsubstituted phenyl group, and σZ is the sub-stituent constant for each substituent used. The
proportionality constant ρ relates
the substituent constant (electron donating or withdrawing) and the
substituent’s effect on rate. It gives information about the type and extent of
charge devel-opment in the activated complex. It is determined by plotting log(kz/ ko) versus σz for a series of substituents. The slope of the linear plot is ρ and is termed the reaction constant. For example, the reaction shown above is an
elimination reaction in which a proton and the nosylate group are eliminated
and a C–N π bond is formed in their place.
The reaction is second order overall, first order in substrate, and first order
in base. The rate constants were measured for several substituted compounds:

These
data were plotted according to the Hammett equation to give the plot in Figure
5.17. The first thing to note is that the Hammett plot is linear. The linearity
of the plot implies that the substituent constants determined for the
ionization of
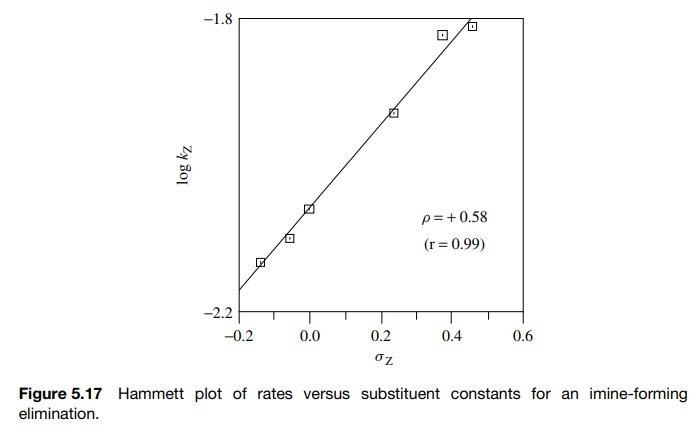
Next
the sign and absolute magnitude of the ρ
value determined from a Ham-mett plot give information about charge development
at the transition state. The sign of ρ
tells whether a positive or negative charge is being developed in the activated
complex relative to the reactants. A positive ρ value means that elec-tron density is increased (negative charge
is being produced) in the activated complex. A negative ρ value means that electron deficiency is being produced (often a
positive charge) in the activated complex. Generally ρ values have abso-lute magnitudes between 0 and 3, but values as
high as 10 or 12 are known. A value of ρ
= 0 means that
substituents have no electronic effect on the reaction rate, and thus no charge
is being developed at the transition state. Large absolute values of ρ mean that substituents influence the
rate greatly, and thus the amount of charge developed in the activated complex
is large and influenced significantly by the electronic properties of the
substituents. For example,
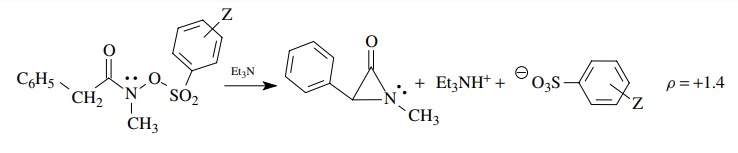
This
ρ = 1.4 means that negative charge is being developed on the
arenesul-fonate group at the transition state, consistent with this group
departing as an anionic leaving group as part of the rate-determining step. The
magnitude of 1.4 means that a significant amount of charge is developed on the
leaving group. This interpretation is possible only by comparing ρ = +1.4
with ρ values for sulfonate leaving
groups in other reactions in which a range of normal values is known. The
normal range is about +0.8
to +1.65; therefore the
value of 1.4 indicates significant charge development at the transition state.
In addition to the positive
Moreover, there is a primary
kinetic deuterium isotope effect for the benzylic position. These observations
allow a detailed structure to be drawn of the activated complex in which
concerted 1,3 elimination results in formation of the three-membered ring product.
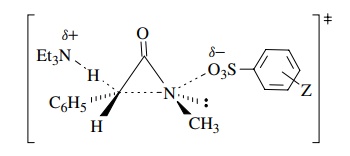
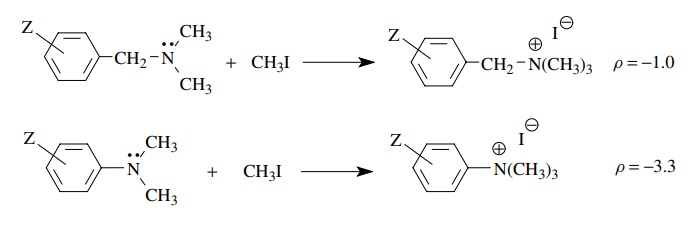
The
reaction of N ,N -dimethylbenzylamines with methyl iodide is found to have ρ = −1.0.
The negative sign indicates that partial positive charge is being produced in
the transition state. The modest size of ρ
is consistent with the charge being developed on the nitrogen, which is
insulated from the aromatic ring by the saturated CH2 group. The
ability of substituents to influence rate is reduced by the insulating
methylene group, and therefore ρ is
smaller. In comparison the methylation of N
,N -dimethylaniline has ρ = −3.3
because the nitrogen atom, on which positive charge is developed in the
transition state, is directly attached to the phenyl ring and the substituents
have a greater influence on the stability of the charge being developed. Hence
the magnitude of ρ is much larger.
For
similar reactions, comparison of the ρ
values can be used to determine which reaction has a greater charge
development. Comparison of the olefin-forming eliminations below reveals which
reaction has greater charge develop-ment at the benzylic position and thus
which has a greater degree of proton removal in the activated complex.
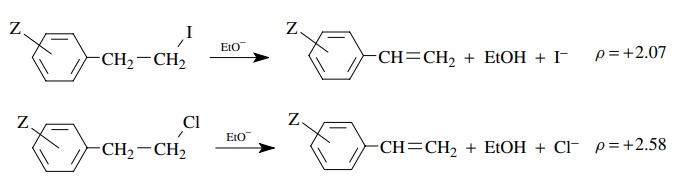
The
larger value of ρ = 2.58 for the chloride leaving group than for the iodide leaving
group (ρ = 2.07) suggests that
greater negative charge is developed on the benzylic position when chloride is
the leaving group than when iodide is the leaving group. Base removes the
proton to a greater extent for the poorer chloride leaving group than for the
better iodo leaving group. Greater electron density on the benzylic position is
required to expel the chloride leaving group than the iodide leaving group
(i.e., chloride needs a greater “push” than iodide). The transition state for
proton removal is later with the chloride leaving group and earlier with the
iodide leaving group. These differences in ρ
values are thus very useful for discussing changes in the structures of
activated complexes and reaction mechanisms caused by structural changes in the
reactants.
Sometimes
Hammett plots of rates versus σz
values are nonlinear. When this occurs, it usually indicates that the model
reaction from which σz
values were determined (the ionization of benzoic acids) does not accurately
model the elec-tronic changes occurring in the reaction being studied. In most
cases this happens when a positive or negative charge is being developed at a
position where direct resonance interactions with the substituent magnify the
electronic effect of sub-stituents. Usually this happens when charge is
developed at a benzylic position or directly on the aromatic ring.
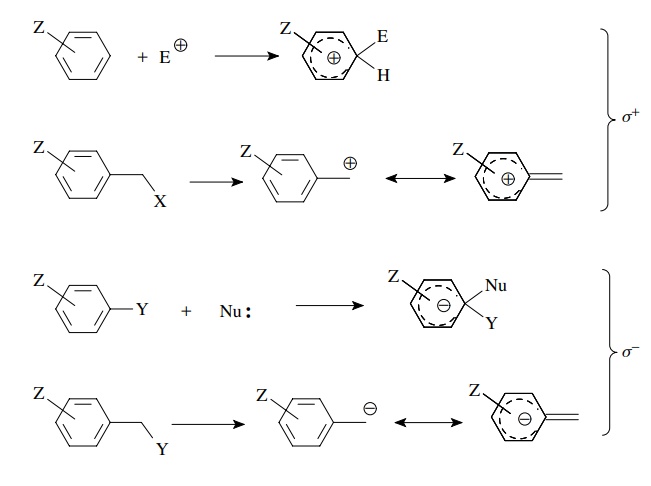
For
these cases, new model reactions were developed and the electronic effects of
substituents were obtained as σ + and σ − substituent constants. The use of σ + constants is applicable for
reactions in which a positive charge is generated on or in direct conjugation
with the aromatic ring, while σ − constants are used
for reactions in which a negative charge is generated on or in direct
conjugation with the phenyl ring. A listing of these constants is found in
Table 5.1. These constants are used in the same way as σz constants. Rates are measured and correlated with the
appropriate set of σ values (σ, σ +, or σ −)
in typical Hammett fashion:
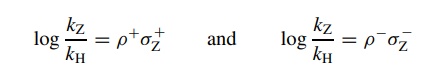
As
before, the σ constants are
positionally dependent, that is, σ + meta is different
than σ + para.
A
better linear correlation of the rates with a particular set of σ values means that the set of σ values used is a better model for the
electronic character of the reaction being studied. Knowing that a better
correlation is found for σ + than
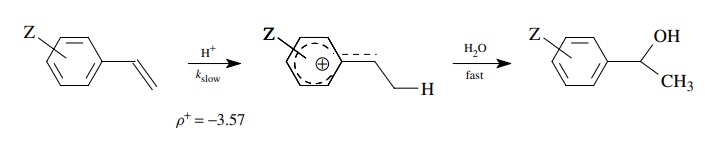
For example, the hydration of
substituted styrenes gives a much better linear correlation of log(kZ/ kH) versus σ + than σ . This is consistent with the rate-determining step being
protonation of the double bond, giving a benzylic cation. At the transition
state, resonance delo-calization of the developing positive charge into the
ring accounts for the better correlation with σZ+
. The value ρ+ = −3.57 means that a significant amount of
positive charge is developed at the transition state.
The
rates of rearrangement of aromatic amine oxides is found to be correlated much
better by σ − than with σ and has ρ− = 3.6. A mechanism consistent with this finding has the oxygen
attacking the substituted aromatic ring, thus increasing the electron density
on the ring. This charge is delocalized over the aromatic ring by resonance and
thus is in direct resonance interaction with substituents in the para position.
The process is much better modeled by σ
− than by σ constants.
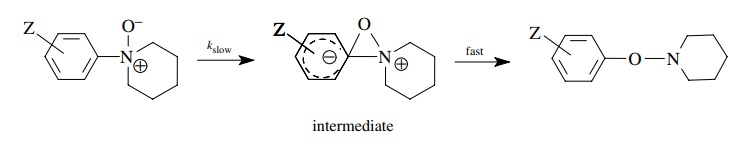
Because
of the way in which they were developed, only σp−
substituent constants are available. If a substituent is meta, then normal σm values are used because
there is no direct resonance interaction with a substituent in the meta
position.
By
a variety of techniques, it is possible to gain detailed insight into the
structure and properties of the activated complex. This is a remarkable ability
considering that the lifetime of the activated complex is on the order of a bond
vibration and thus cannot be observed directly. This ability to determine the
energy of the transition state and structurally characterize the activated
complex is a cornerstone of organic chemistry. It allows us to determine
mechanisms of reactions, provides a way to evaluate and predict the effects of
structural changes on the reactivity of molecules, and enables us to devise new
reactions and processes based on new mechanistic principles that are
discovered. The result is an ever-increasing arsenal of reactions than can be
used most effectively for carrying out needed organic transformations.
Related Topics
