Hypolipidaemic Drugs
| Home | | Pharmacology |Chapter: Essential pharmacology : Hypolipidaemic Drugs And Plasma Expanders
The hypolipidaemic drugs have attracted considerable attention because of their potential to prevent cardiovascular disease by retarding the accelerated atherosclerosis in hyperlipidaemic individuals.
HYPOLIPIDAEMIC DRUGS
These are drugs which
lower the levels of lipids and lipoproteins in blood.
The hypolipidaemic
drugs have attracted considerable attention because of their potential to
prevent cardiovascular disease by retarding the accelerated atherosclerosis in
hyperlipidaemic individuals.
Lipid Transport
Lipids are carried in
plasma in lipoproteins after getting associated with several apoproteins;
plasma lipid concentrations are dependent on the concentration of lipoproteins.
The core of lipoprotein globules consists of triglycerides (TGs) or cholesteryl
esters (CHEs) while the outer polar layer has phospholipids, free cholesterol
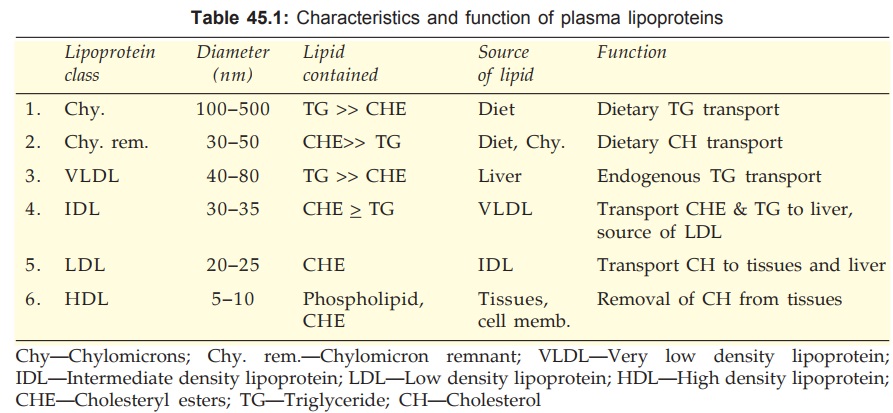
(CH) and apoproteins. The lipoproteins have been divided into 6 classes on the
basis of their pCh. No. size and density. They also differ in the nature of
apoproteins, the ratio of TG and CHE, tissue of origin and fate. These are
given in Table 45.1.
Dietary lipids are
absorbed in the intestine with the help of bile acids. Chylomicrons (Chy) are
formed and passed into lacteals—reach blood stream via thoracic duct. During their passage through capillaries, the
endothelium bound lipoprotein lipase hydrolyses the TGs into fatty acids which pass
into muscle cells to be utilized as energy source and in fat cells to be
reconverted into TGs and stored. The remaining part—chylomicron remnant (Chy.
rem.) containing mainly CHE and little TG is engulfed by liver cells, which
have receptors for the surface apoproteins of Chy. rem., and digested. Free CH
that is liberated is either stored in liver cells after re-esterification or
incorporated into a different lipoprotein and released in blood or excreted in
bile as CH/bile acids.
Liver secretes very low density lipoproteins (VLDL)
containing mainly TG and some CHE into blood. VLDL is acted upon by endothelial
lipoprotein lipase in the same way as on Chy and the fatty acids pass into
adipose tissue and muscle; the remnant called intermediate density lipoprotein
(IDL) now contains more CHE than TG. About half of the IDL is taken back by the liver cells by attachment to
another receptor (LDL receptor), while the rest loses the remaining TGs
gradually and becomes low density lipoprotein (LDL) containing only CHE.
The LDL circulates in plasma for a
long time; its uptake into liver and other tissues is dependent on the need for
CH. NO. The rate of LDL uptake is regulated by the rate of LDL receptor
synthesis in a particular tissue.
The CHE of LDL is
deesterified and used mainly for cell membrane formation. The CH released into
blood from degradation of membranes is rapidly incorporated in high density
lipoproteins (HDL), esterified with the help of an enzyme lecithin: cholesterol acyltransferase (LCAT) and
transferred back to VLDL or IDL, completing the cycle.
The excess
lipoproteins in plasma are phagocytosed by macrophages for disposal. When too
much of lipoproteins have to be degraded in this manner, CH is deposited in atheromas (in arterial walls) and xanthomas (in skin and tendons). Raised levels of VLDL, IDL and
LDL (rarely Chy and Chy. rem. also) are atherogenic, while HDL may be protective,
because HDL facilitates removal of CH from tissues.
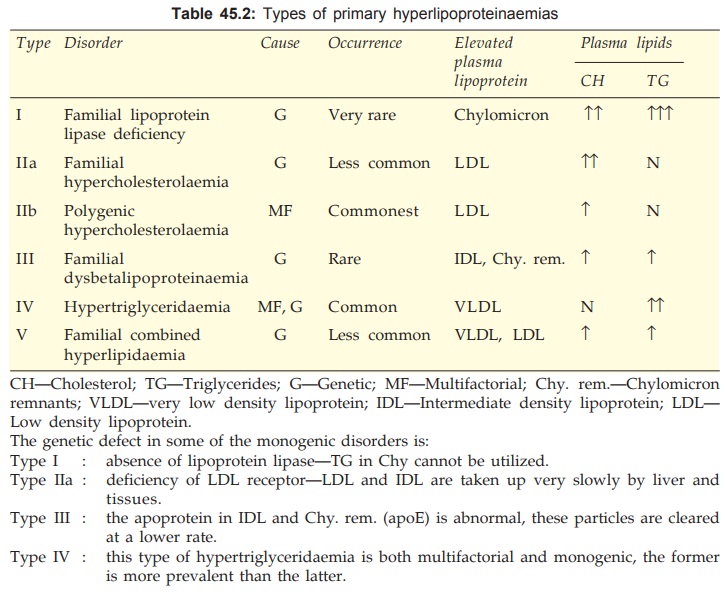
Hyperlipoproteinaemias can be:
Secondary: associated with
diabetes, myxoedema, nephrotic syndrome, chronic alcoholism, drugs
(corticosteroids, oral contraceptives, β blockers) etc.
Primary: due to:
·
A single gene defect: is familial and called
‘monogenic’ or genetic.
· Multiple genetic, dietary and physical activity
related causes: ‘polygenic’ or multifactorial.
On the whole, LDL is the primary carrier of plasma CHE, and VLDL
that of TGs. The important features of major types of hyperlipoproteinaemias
are given in Table 45.2.
Classification
1.
HMGCoA reductase inhibitors (Statins):
Lovastatin, Simvastatin, Pravastatin, Atorvastatin, Rosuvastatin
2.
Bile acid sequestrants (Resins):
Cholestyramine, Colestipol
3.
Activate lipoprotein lipase (Fibric acid derivatives):
Clofibrate,
Gemfibrozil, Bezafibrate, Fenofibrate.
4.
Inhibit lipolysis and triglyceride
synthesis:
Nicotinic
acid
5.
Others:
Ezetimibe, Gugulipid.
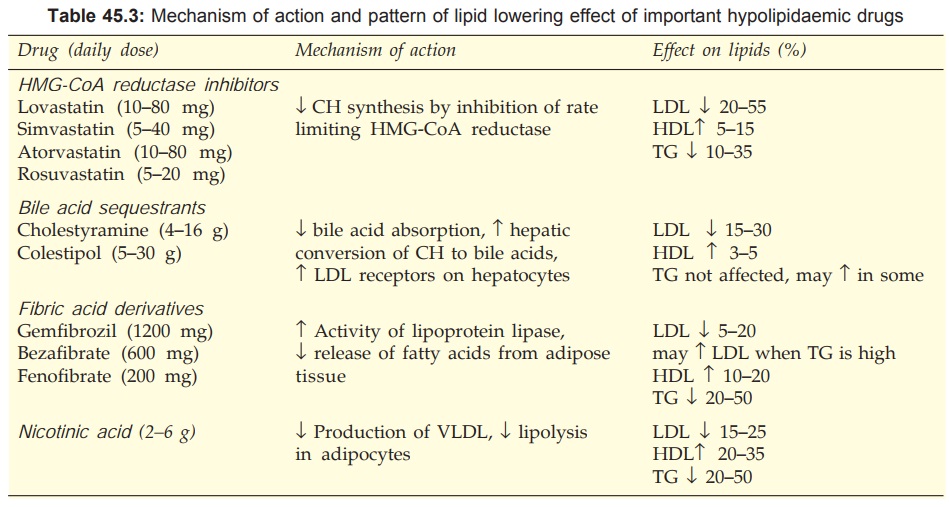
The mechanism of action and profile of lipid lowering effect of
important hypolipidaemic drugs is summarized in Table 45.3.
HMG-CoA REDUCTASE INHIBITORS (STATINS)
Introduced in the
1980s, this class of compounds are the most efficacious and best tolerated hypolipidaemic
drugs. They competitively inhibit conversion of 3Hydroxy3methyl glutaryl
coenzyme A (HMG-CoA) to mevalonate (rate limiting step in CH synthesis) by the
enzyme HMG-CoA reductase. Therapeutic doses reduce CH synthesis by 20–50%. This
results in compensatory increase in LDL receptor expression on liver cells → increased receptor
mediated uptake and catabolism of IDL and LDL. Over long-term, feedback
induction of HMG-CoA reductase tends to increase CH synthesis, but a steady state
is finally attained with a dose-dependent lowering of LDL-CH levels.
Different statins differ
in their potency and maximal efficacy in reducing LDL-CH. NO. The daily dose
for lowering LDL-CH by 30–35% is lovastatin 40 mg, pravastatin 40 mg, simvastatin
20 mg, atorvastatin 10 mg, rosuvastatin 5 mg. Moreover, at their maximum
recommended doses simvastatin, atorvastatin and rosuvastatin can reduce LDL-CH
by upto 45–55%, while the ceiling effect of lovastatin and pravastatin is 35–40%
LDL-CH reduction. All statins produce peak LDL-CH lowering after 1–2 weeks
therapy. Hepatic synthesis of VLDL is concurrently reduced and its removal from
plasma is enhanced.
A dose-dependent effect is seen with all statins. With lovastatin a mean reduction of LDL-CH by 25% at 20 mg/day, 32% at 40 mg/day and 40% at 80 mg/day has been measured. A concurrent fall by 10–30% in plasma TG level, probably due to reduction of VLDL occurs. A rise in HDLCH by 5–15% is also noted. Simultaneous use of bile salt sequestrant augments the LDL lowering effect upto 60% and addition of nicotinic acid to this combination may boost the effect to 70% reduction in LDL-CH. NO. Statins are effective in secondary hyper-cholesterolaemias also. The more efficacious statins (simvastatin, atorvastatin, rosuvastatin) given at their higher doses effectively reduce TGs (by 25% to 35%) when they are moderately raised, but not when they are markedly raised.
Because HMG-CoA reductase activity is maximum at midnight, all
statins are administered at bed time to obtain maximum effectiveness. However,
this is not necessary for atorvastatin and rosuvastatin, which have long plasma
t½.
All statins, except rosuvastatin are metabolized primarily by
CYP3A4. Inhibitors and inducers of this isoenzyme respectively increase and
decrease statin blood levels.
Lovastatin It is the first
clinically used statin; is lipophilic and given
orally in the precursor lactone form. Absorption is incomplete and first pass
metabolism is extensive. Metabolites are excreted mainly in bile. The t½ is
short (1–4 hours).
Dose: 10–40 mg/day (max. 80
mg).
ROVACOR, AZTATIN,
LOVAMEG 10, 20 mg tabs.
Simvastatin It is twice as potent as lovastatin; also more efficacious. A greater rise in HDLCH
(when low) has been noted with simvastatin than others. Like lovastatin, it is
lipophilic and given in the lactone precursor form. Oral absorption is better
and first pass metabolism extensive; t½ is 2–3 hr.
Dose: 5–20 mg/day (max. 40
mg)
SIMVOTIN, SIMCARD,
ZOSTA 5, 10, 20 mg tabs.
Pravastatin It is hydrophilic and given in the active form. At low doses it is equipotent to
lovastatin, but at higher doses (40–80 mg/day), CH lowering effect is less. It
can be employed when reduction of LDL-CH by < 25% is contemplated. An
additional action of decrease in plasma fibrinogen level has been observed. The
t½ is 1–3 hours.
PRAVATOR 10, 20 mg
tabs.
Atorvastatin This newer statin is more potent and appears to have the highest LDL-CH
lowering efficacy at maximal daily dose of 80 mg. At this dose a greater
reduction in TGs is noted if the same was raised at baseline. Atorvastatin has
a much longer plasma t½ of 18–24 hr than other statins, and has additional
antioxidant property.
Dose: 1040 mg/day (max. 80 mg)
AZTOR, ATORVA, ATORLIP
10, 20 mg tabs.
Rosuvastatin This is the latest and the most potent statin (10 mg rosuvastatin ~ 20
mg atorvastatin), with a plasma t½ of 18–24 hours. Greater LDL-CH reduction can
be obtained in severe hypercholesterolaemia; partly due to its longer
persistence in the plasma. In patients with raised TG levels, rosuvastatin
raises HDLCH by 15–20% (greater rise than other statins).
Dose: Start with 5 mg OD,
increase if needed upto 20 mg/ day,
(max 40 mg/day)
ROSUVAS, ROSYN 5, 10,
20 mg tabs.
Adverse Effects
All statins are
remarkably well tolerated; overall incidence
of side effects not differing from placebo. Notable side effects are:
·
Headache, nausea, bowel upset, rashes.
·
Sleep disturbances (probably more with
lipophilic drugs).
·
Rise in serum transaminase can occur, but
liver damage is rare.
·
Muscle tenderness and rise in CPK levels occurs
infrequently. Myopathy is the only serious reaction, but is rare (< 1 per
1000). Few fatalities due to rhabdomyolysis are on record. Myopathy is more common
when nicotinic acid/gemfibrozil or CYP3A4 inhibitor— ketoconazole/
erythromycin/cyclosporine/ HIV protease inhibitor is given concurrently.
Gemfibrozil inhibits the hepatic uptake of statins by the organic anion
transporter OATP2. Fenofibrate interferes the least with statin
uptake/metabolism and should be preferred for combining with them.
Use
Statins are the first choice drugs for primary hyperlipidaemias with raised LDL and total CH
levels, with or without raised TG levels (Type IIa, IIb, V), as well as for
secondary (diabetes, nephrotic syndrome) hyper-cholesterolaemia.
Efficacy of statins in reducing raised LDL-CH associated
mortality and morbidity is now well established.
In the ‘Scandinavian Simvastatin Survival Study’ (4S study, 1994),
patients with history of MI (80%) or angina (20%) and raised serum CH level (>
212 mg/dl) were treated with simvastatin or placebo. Simvastatin reduced total
CH by 25%, LDL-CH by 35%, raised HDLCH by 8%. Over a period of 6 years coronary
artery disease (CAD) mortality was less by 42%, overall mortality by 30% and
cerebrovascular events by 30% in the simvastatin group. Similar results have
been obtained with other statins, e.g. the West of Scotland Coronary Prevention
Study (WOSCOPS) in men with no history of MI has found pravastatin to lower
risk of MI by 31% and all cause mortality by 22%.
Subsequent studies like Long-term intervention with pravastatin
in ischaemic disease (LIPID1998), Airforce/ Texas coronary atherosclerosis
prevention study (AFCAPS/TexCAPS1998), Cholesterol and recurrent events (CARE1998),
and trials conducted by Heart Protection Study Collaborative Group (2002, 2004)
in over 20,000 patients have confirmed the mortality and morbidity benefits of
statins, including stroke prevention.
Beneficial effects in
subjects who have raised CH levels but no evidence of CAD may relate to
improved coronary artery compliance and atheromatous plaque stabilization due
to suppression of macrophage mediated inflammation, reducing chances of plaque
rupture and thrombus formation. Improvement in endothelial function due to
increased NO production and reduction in LDL oxidation are proposed as
additional mechanisms by which statins may exert anti-atherosclerotic action.
On the basis of these results as well as the excellent patient acceptability,
the statins are being increasingly used for primary and secondary hyper-cholesterolaemia
with or without raised TG levels. They are the first choice drugs for
dyslipidaemia in diabetics.
BILE ACID SEQUESTRANTS (RESINS)
Cholestyramine and Colestipol
These are basic ion exchange resins supplied in the chloride form.
They are neither digested nor absorbed in the gut: bind bile acids in the
intestine interrupting their enterohepatic circulation. Faecal excretion of
bile salts and CH (which is absorbed with the help of bile salts) is increased.
This indirectly leads to enhanced hepatic metabolism of CH to bile acids. More
LDL receptors are expressed on liver cells: clearance of plasma IDL, LDL and
indirectly that of VLDL is increased.
Resins have been shown
to retard atherosclerosis, but are not popular clinically because they are
unpalatable, inconvenient, have to be taken in large doses, cause flatulence
and other g.i. symptoms, interfere with absorption of many drugs and have poor patient
acceptability.
FIBRIC ACID DERIVATIVES
The fibrates
(isobutyric acid derivatives) primarily activate lipoprotein lipase which is a
key enzyme in the degradation of VLDL resulting in lowering of circulating TGs.
This effect is exerted through paroxisome proliferator-activated receptor α (PPARα) that is a gene transcription
regulating receptor expressed in liver, fat and muscles. Activation of PPARα enhances lipoprotein
lipase synthesis and fatty acid oxidation. PPARα may also mediate
enhanced LDL receptor expression in liver seen particularly with second
generation fibrates. Fibrates decrease hepatic TG synthesis as well. A
peripheral effect reducing circulating free fatty acids has also been shown.
Drugs in this class
primarily lower TG levels by 20–50%, generally accompanied by 10–15% decrease
in LDL-CH and a 10–15% increase in HDLCH. NO. In some patients with hyper-triglyceridaemia
LDL-CH may rise, partly because of inability of LDL receptor to clear the
excess number of LDL pCh. No.s generated by enhanced VLDL catabolism. The increase
in HDLCH is at least in part due to transfer of surface lipid components from
catabolized VLDL to HDL, and partly due to increased production of HDL
apoproteins (apo AI, apo AII) by liver. Gemfibrozil also appears to reduce VLDL
secretion by liver.
LDL composition may be
altered. Gemfibrozil and bezafibrate have been shown to shift small dense LDL pCh.
No.s (believed to be more atherogenic) to larger less dense pCh. No.s.
Clofibrate
It was a widely used
hypolipidaemic drug, but later evidence
showed that it does not prevent atherosclerosis, therefore has gone out of use.
Gemfibrozil
This fibric acid
derivative effectively lowers plasma TG level by enhancing breakdown and
suppressing hepatic synthesis of TGs. Besides high efficacy in type III hyperlipoproteinemia,
gemfibrozil has shown action in subjects with raised blood CH in addition. In
the ‘Helsinki Heart Study’ men without known CAD treated with gemfibrozil had a
34% reduction in fatal and nonfatal MI, though overall mortality was not affected.
Additional actions to decrease the level of clotting factor VII-phospholipid
complex and promotion of fibrinolysis have been observed, which may contribute
to the anti-atherosclerotic effect.
Pharmacokinetics
Gemfibrozil is
completely absorbed orally, metabolized
by glucuronidation and undergoes some enterohepatic circulation. It is excreted
in urine; elimination t½ 1–2 hr.
Adverse Effects
Common side effects
are epigastric distress,
loose motions.
Skin rashes, body
ache, eosinophilia, impotence, headache and blurred vision have been reported.
Myopathy is uncommon. Gemfibrozil + statin increases risk of myopathy.
Incidence of gallstone
is not increased as seen with clofibrate.
It is contraindicated during pregnancy.
GEMPAR, NORMOLIP 300
mg cap. LOPID 300 mg cap, 600 mg and 900 mg tabs.
Use
In a dose of 600 mg BD
taken before meals, gemfibrozil is the
drug of choice for patients with markedly raised TG levels, whether or not CH
levels are also raised. Episodes of acute pancreatitis are prevented in patients
with chylomicronaemia and severe hyper-triglyceridaemia. It is most effective
in type III hyper-lipoproteinaemia; also a first line drug in type IV and type
V disease. It may be used as an adjuvant drug in type IIb patients.
Bezafibrate
This second generation
fibric acid derivative is an
alternative for gemfibrozil in mixed hyper-lipidaemias (type III, IV and V).
Though it has also been indicated in hyper-cholesterolaemia (type II), it is
inferior to statins and resins. It has not shown propensity to increase LDL-CH
in hyper-triglyceridaemic patients and appears to have greater LDL-CH lowering
action than gemfibrozil. Decreased level of circulating fibrinogen and glucose
has been demonstrated. The 5 year ‘Bezafibrate Coronary Atherosclerosis Intervention
Trial’ (BECAIT) in young male post-MI subjects has shown it to slow
atherosclerotic process and reduce coronary events.
Adverse effects and
contraindications of bezafibrate are similar to other fibrates. Main side
effects are g.i. upset, myalgia, rashes. Dose reduction is needed in elderly
and in renal insufficiency. Action of oral anticoagulants may be enhanced.
In contrast to other
fibrates, combination of bezafibrate with a statin has not so far been found to
increase the incidence of rhabdomyolysis.
Dose: 200 mg TDS with meals.
BEZALIP 200, 400 mg
tab.
Fenofibrate
Another 2nd generation
prodrug fibric acid derivative
which has greater HDL–CH raising and greater LDL-CH lowering action than other
fibrates: may be more appropriate as an adjunctive drug in subjects with raised
LDL-CH levels in addition to raised TG levels. No rise in LDL-CH has been
observed in patients with high TG levels. Its t½ is 20 hr. Adverse effects are
myalgia, hepatitis, rashes. Cholelithiasis and rhabdomyolysis are rare. Fenofibrate
appears to be the most suitable fibrate for combining with statins, because
statin metabolism is minimally affected and enhancement of statin myopathy risk
is lower. Indications of fenofibrate are similar to that of gemfibrozil.
Dose: 200 mg OD with meals.
FENOLIP, LIPICARD 200
mg cap.
NICOTINIC ACID (NIACIN)
It is a B group
vitamin (see Ch. No. 67) which in
much higher doses reduces plasma lipids. This action is unrelated to its
vitamin activity and not present in nicotinamide. When nicotinic acid is given,
TGs and VLDL decrease rapidly, followed by a modest fall in LDL-CH and total CH.
A 20–50% reduction in plasma TGs and 15–25% reduction in CH levels has been
recorded. Nicotinic acid is the most effective drug to raise HDL-CH; a 20–35% increase
is generally obtained. Relatively lower dose suffices to raise HDL–CH.
Nicotinic acid reduces
production of VLDL in liver by inhibiting TG synthesis. Indirectly the VLDL
degradation products IDL and LDL are also reduced. No direct effect on CH and
bile acid metabolism has been found. It inhibits intracellular lipolysis in
adipose tissue and increases the activity of lipoprotein lipase that clears
TGs.
A cell surface G-protein
coupled receptor which negatively regulates adipocyte adenylyl cyclase has been
found to selectively bind nicotinic acid, and has been called ‘niacin
receptor’. Nicotinic acid appears to inhibit lipolysis in adipose tissue by
decreasing hormone stimulated intracellular cAMP formation through this
receptor. Hepatic VLDL production is believed to be decreased due to reduced
flow of fatty acids from adipose tissue to liver.
Adverse Effects
The large doses needed
for hypolipidaemic action
are poorly tolerated. Only about half of the patients are able to take the full
doses.
Nicotinic acid is a
cutaneous vasodilator: marked flushing, heat and itching (especially in the
blush area) occur after every dose. This can be minimized by starting with a
low dose taken with meals and gradually increasing as tolerance develops.
Aspirin taken daily largely prevents the reaction (PGs may be involved).
Dyspepsia is very
common; vomiting and diarrhoea occur when full doses are given. Peptic ulcer
may be activated.
Dryness and
hyperpigmentation of skin can be troublesome. Other long-term effects are:
Liver dysfunction and
jaundice. Serious liver damage is the most important risk.
Hyperglycaemia, precipitation
of diabetes (should not be used in diabetics) . Hyperuricaemia and gout, atrial
arrhythmias. It is contraindicated during pregnancy and in children.
Interaction
Postural hypotension
may occur in patients on
antihypertensives when they take nicotinic acid.
Risk of myopathy due
to statins is increased.
Dose: Start with 100 mg TDS,
gradually increase to 2–6 g per day in divided
doses. It should be taken just after food to minimize flushing and itching.
NIALIP 250, 375, 500
mg tabs.
Use
Nicotinic acid is a
wide spectrum hypolipidaemic drug. It is highly efficacious in hyper-triglyceridaemia
(type III, IV, V) whether associated with raised CH level or not. It is mostly
used to lower VLDL and raise HDL levels, and as an adjunctive drug to statins/fibrates.
Nicotinic acid is the
most effective drug in reducing plasma TG levels and controlling pancreatitis
in genetic type IV and type V disorders. Long-term use prevents further attacks
of pancreatitis. Given over long-term in postMI patients, it has been found to
reduce recurrences of MI and overall mortality. However, because of marked side
effects, use of nicotinic acid is restricted to high risk cases only.
OTHER HYPOLIPIDAEMICS
Ezetimibe
It is a new drug of
its own kind that acts by inhibiting
intestinal absorption of cholesterol and phytosterols. It interferes with a
specific CH transport protein NPC1C1 in the intestinal mucosa and reduces absorption
of both dietary and biliary CH. NO. There is compensatory increase in hepatic
CH synthesis, but LDL-CH level is lowered by 15–20%. The enhanced CH synthesis
can be blocked by statins, and the two drugs have synergistic LDL-CH lowering
effect.
Due to very poor aqueous
solubility, ezetimibe is not absorbed as suCh. No. It is absorbed partly after
getting conjugated with glucuronic acid in the intestinal mucosa → secreted in bile → undergoes enterohepatic
circulation and is mainly excreted in faeces. A plasma t½ of 22 hours has been
calculated.
Used alone, ezetimibe
is a weak hypo-cholesterolaemic drug; LDL CH lowering beyond 15–20% is not
obtained by increasing the dose. Though it may be used alone in mild hyper-cholesterolaemia
when a statin is contraindicated/ not tolerated, its main value is to
supplement statins without increasing their dose. The combination of ezetimibe
+ low dose of a statin is as effective in lowering LDL-CH as high dose of
statin alone. Upto 60% decrease in LDL-CH level has been obtained with a
combination of simvastatin + ezetimibe. No specific adverse effect, except reversible
hepatic dysfunction and rarely myositis has been noted with ezetimibe.
Dose: 10 mg OD; ZETICA, EZEDOC 10 mg
tab.
Gugulipid
It is a mixture of
sterones obtained from ‘gum guggul’ which has
been used in Ayurveda. Modest lowering of plasma CH and TGs occurs after
continued use of gugulipid. It is well tolerated: loose stools are the only
significant side effect.
Related Topics
