Oral Hypoglycaemic Drugs
| Home | | Pharmacology |Chapter: Essential pharmacology : Insulin, Oral Hypoglycaemic Drugs and Glucagon
These drugs lower blood glucose levels and are effective orally. The chief draw back of insulin is—it must be given by injection. Orally active drugs have always been searched.
ORAL HYPOGLYCAEMIC DRUGS
These drugs lower
blood glucose levels and are effective orally. The chief draw back of insulin
is—it must be given by injection. Orally active drugs have always been
searched.
The early sulfonamides
tested in 1940s produced hypoglycaemia as side effect. Taking this lead, the
first clinically acceptable sulfonylurea tolbutamide
was introduced in 1957. Others followed soon after. In the 1970s many so called
‘second generation’ sulfonylureas have been developed which are 20–100 times
more potent. Clinically useful biguanide phenformin
was developed parallel to sulfonylureas in 1957. Recently 3 newer classes of drugs,
viz. α glucosidase inhibitors, meglitinide analogues and thiazolidinediones have been inducted.
SULFONYLUREAS
First generation
Tolbutamide
Chlorpropamide
Second generation
Glibenclamide
(Glyburide)
Glipizide
Gliclazide
Glimepiride
BIGUANIDES
Metformin
MEGLITINIDE / PHENYL ALANINE
ANALOGUES
Repaglinide
Nateglinide
THIAZOLIDINEDIONES
Rosiglitazone
Pioglitazone
α GLUCOSIDASE INHIBITORS
Acarbose
Miglitol
SULFONYLUREAS
The
generic formula of sulfonylureas is—
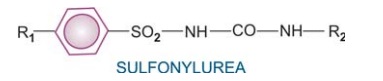
All
have similar pharmacological profile—sole significant action being lowering of
blood glucose level in normal subjects and in type 2 diabetics, but not in type
1 diabetics.
Mechanism Of Action
Sulfonylureas
provoke a brisk release of
insulin from pancreas. They act on the so called ‘sulfonylurea receptors’
(SUR1) on the pancreatic β cell membrane—cause depolarization by
reducing conductance of ATP sensitive K+ channels. This enhances Ca2+ influx degranulation.
The rate of insulin secretion at any glucose concentration is increased. In
type 2 DM the kinetics of insulin release in response to glucose or meals is
delayed and subdued. The sulfonylureas primarily augment the 2nd phase insulin
secretion with little effect on the 1st phase. That they do not cause
hypoglycaemia in pancreatectomised animals and in type 1 diabetics (presence of
at least 30% functional β cells is essential for their action) confirms
their indirect action through pancreas.
A
minor action reducing glucagon secretion, probably by increasing insulin and
somatostatin release has been demonstrated. Hepatic degradation of insulin is
slowed.
Extrapancreatic Action After chronic administration, the insulinaemic
action of sulfonylureas declines probably due to down regulation of
sulfonylurea receptors on β cells, but improvement in glucose tolerance
is maintained. In this phase, they sensitize the target tissues (especially
liver) to the action of insulin. This is due to increase in number of insulin
receptors and/or a postreceptor action—improving translation of receptor
activation. It is hypothesized that long term improvement in carbohydrate tolerance
leads to a decreased insulin concentration in blood which reverses the down
regulation of insulin receptors—apparent increase in their number. A direct
extrapancreatic action of sulfonylureas to increase insulin receptors on target
cells and to inhibit gluconeogenesis in liver has been suggested, but appears
to have little clinical relevance.
Pharmacokinetics
All sulfonylureas are
well absorbed orally, and
are 90% or more bound to plasma proteins: have low volumes of distribution
(0.2–0.4 L/kg). Some are primarily metabolized—may produce active metabolite;
others are mainly excreted unchanged in urine. Accordingly they should be used
cautiously in patients with liver or kidney dysfunction.
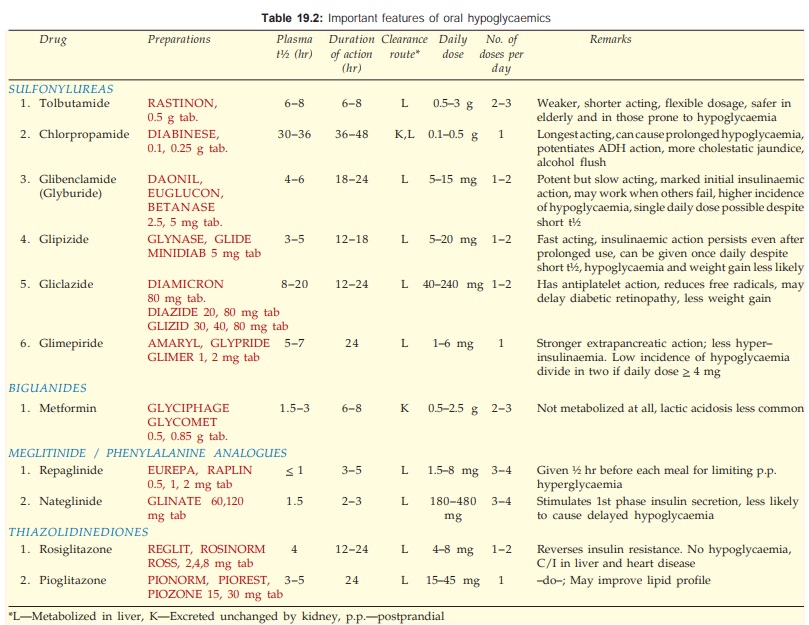
The
distinctive features of different sulfonylureas are given in Table 19.2.
Interactions
Drugs that enhance sulfonylurea action (may precipitate
hypoglycaemia) are—
a. Displace from protein binding: Phenylbutazone,
sulfinpyrazone, salicylates, sulfonamides, PAS.
b. Inhibit metabolism/excretion: Cimetidine, sulfonamides,
warfarin, chloramphenicol, acute alcohol intake (also synergises by causing
hypoglycaemia).
c. Synergise with or prolong pharmacodynamic
action: Salicylates, propranolol (cardioselective β1 blockers less liable),
sympatholytic antihypertensives, lithium, theophylline, alcohol (by inhibiting gluconeogenesis).
Drugs that decrease sulfonylurea action (vitiate diabetes
control) are—
a. Induce metabolism: Phenobarbitone,
phenytoin, rifampicin, chronic
alcoholism.
b. Opposite action/suppress insulin release: Corticosteroids,
diazoxide, thiazides, furosemide, oral contraceptives.
Adverse Effects
Incidence
of adverse effects is quite low (3–7%).
1. Hypoglycaemia It is the commonest
problem, may occasionally be
severe and rarely fatal. It is more common in elderly, liver and kidney disease
patients and when
potentiating drugs are added. Chlorpropamide is a frequent culprit due to its
long action. Tolbutamide carries lowest risk due to its low potency and short
duration of action. Lower incidence is also reported with glipizide,
glibenclamide, glimepiride.
Treatment is to give
glucose, may be for a few days because hypoglycaemia may recur.
2. Nonspecific side effects Nausea, vomiting, flatulence, diarrhoea
or constipation, headache, paresthesias and weight gain.
3. Hypersensitivity Rashes, photosensitivity, purpura, transient leukopenia, rarely agranulocytosis.
Chlorpropamide in
addition causes cholestatic jaundice, dilutional hyponatremia (sensitises the
kidney to ADH action), intolerance to alcohol in predisposed subjects (flushing
and a disulfiram like reaction); other sulfonylureas are less prone to this
interaction.
Tolbutamide reduces
iodide uptake by thyroid but hypothyroidism does not occur.
Safety of
sulfonylureas during pregnancy is not established—change over to insulin. They
are secreted in milk: should not be given to nursing mothers.
BIGUANIDES
Two biguanide
antidiabetics, phenformin and metformin were introduced in the 1950s.
Because of higher risk of lactic
acidosis, phenformin was withdrawn in many countries and has been banned in
India since 2003.
The generic formula of
biguanides is:
They differ markedly
from sulfonylureas: cause little or no hypoglycaemia in nondiabetic subjects,
and even in diabetics episodes of hypoglycaemia due to metformin are rare. They
do not stimulate pancreatic β cells. Metformin is reported to improve lipid
profile as well in type 2 diabetics.
Mechanism Of Action
It is not clearly
understood. Biguanides do not cause insulin release, but presence of some
insulin is essential for their action. Explanations offered for their hypoglycaemic
action are—
1) Suppress hepatic
gluconeogenesis and glucose output from liver: the major action.
2) Enhance insulinmediated
glucose disposal in muscle and fat. Though they do not alter translocation of
GLUT4 (the major glucose transporter in skeletal muscle), they enhance GLUT1
transport from intracellular site to plasma membrane. The effect thus differs
from that of insulin.
3) Retard intestinal
absorption of glucose, other hexoses, amino acids and vit B12.
4) Interfere with
mitochondrial respiratory chain—promote peripheral glucose utilization by
enhancing anaerobic glycolysis. However, metformin binds less avidly to
mitochondrial membrane.
Actions 3 and 4 appear to contribute little to the therapeutic
effect.
Pharmacokinetics
The important features
are given in Table 19.2. Clearance
of metformin approximates g.f.r. It accumulates and increases the risk of
lactic acidosis in renal failure.
Adverse Effects
Abdominal pain,
anorexia, nausea, metallic taste,
mild diarrhoea and tiredness are the frequent side effects. Metformin does not
cause hypoglycaemia except in overdose.
Lactic acidosis Small increase in
blood lactate occurs with metformin,
but lactic acidosis is rare (<1 per 10,000 patient years) because it is
poorly concentrated in hepatic cells. Alcohol ingestion can precipitate severe
lactic acidosis.
Vit B12 deficiency due to interference with its absorption can occur with high dose of
metformin.
In addition to general
restrictions for use of oral hypoglycaemics (see below), biguanides are contraindicated in hypotensive states,
cardiovascular, respiratory, hepatic and renal disease and in alcoholics
because of increased risk of lactic acidosis.
MEGLITINIDE / DPHENYLALANINE ANALOGUES
These are recently developed quick and short
acting insulin releases.
Repaglinide
It is a meglitinide analogue oral hypoglycaemic designed to normalise mealtime
glucose excursions. Though not a sulfonylurea, it acts in an analogous mannerby
binding to sulfonylurea receptor as well as to other distinct receptors → closure of ATP
dependent K+ channels → depolarisation → insulin release.
Repaglinide induces rapid onset shortlasting
insulin release. It is administered before each major meal to control
postprandial hyperglycaemia; the dose should be omitted if a meal is missed.
Because of short lasting action it may have a lower risk of serious
hypoglycaemia. Side effects are mild headache, dyspepsia, arthralgia and weight
gain.
Repaglinide is indicated only in type 2 DM as
an alternative to sulfonylureas, or to supplement metformin/longacting insulin.
It should be avoided in liver disease.
Nateglinide
This D-phenylalanine derivative principally stimulates the 1st phase insulin
secretion resulting in rapid onset and shorter duration of hypoglycaemic action
than repaglinide. Ingested 10–20 min before meal, it limits postprandial
hyperglycaemia in type 2 diabetics without producing late phase hypoglycaemia.
There is little effect on fasting blood glucose level. Episodes of
hypoglycaemia are less frequent than with sulfonylureas. Side effects are
dizziness, nausea, flu like symptoms and joint pain. It is used in type 2 DM along
with other antidiabetics, to control postprandial rise in blood glucose.
THIAZOLIDINEDIONES
Two
thiazolidinediones Rosiglitazone and Pioglitazone are available. This novel
class of oral antidiabetic drugs are
selective agonists for the nuclear peroxisome
proliferator activated receptor γ (PPARγ) which enhances the
transcription of several insulin responsive genes. They tend to reverse insulin
resistance by stimulating GLUT4 expression and translocation: entry of glucose
into muscle and fat is improved. Hepatic gluconeogenesis is also suppressed.
Activation of genes regulating fatty acid metabolism and lipogenesis in adipose
tissue contributes to the insulin sensitizing action. Adipocyte turnover and
differentiation may also be affected. Thus, fatty tissue is a major site of
their action. The magnitude of blood glucose reduction is somewhat less than
sulfonylureas and metformin. Improved glycaemic control results in lowering of
circulating HbA1C and insulin levels in type 2 DM patients.
Pioglitazone
lowers serum triglyceride level and raises HDL level without much change in LDL
level, probably because it acts on PPARα as well. The effect
of rosiglitazone on lipid profile is inconsistent.
Both
pioglitazone and rosiglitazone are well tolerated; adverse effects are plasma
volume expansion, edema, weight gain, headache, myalgia and mild anaemia.
Monotherapy with glitazones is not associated with hypoglycaemic episodes. Few
cases of hepatic dysfunction and some cardiovascular events have been reported;
CHF may be precipitated or worsened. Monitoring of liver function is advised.
They are contraindicated in liver disease and in CHF. Rosiglitazone has been
found to increase the risk of fractures, especially in elderly women.
Rosiglitazone
is metabolized by CYP2C8 while pioglitazone is metabolized by both CYP2C8 and
CYP3A4. Failure of oral contraception may occur during pioglitazone therapy.
Ketoconazole inhibits metabolism of pioglitazone. Drug interactions are less
marked with rosiglitazone.
The thiazolidinediones
are indicated in type 2 DM, but not in type 1 DM. They reduce blood glucose and
HbA1c without increasing circulating insulin. Some patients may not
respond (non-responders), especially those with low baseline insulin levels.
Glitazones are primarily used to supplement sulfonylureas/metformin and in case
of insulin resistance. They may also be used as monotherapy (along with diet
and exercise) in mild cases. Reduction in mortality due to myocardial
infarction and stroke (macrovascular complications) has been obtained in type 2
DM.
Several
reports associating precipitation of CHF after combined use of glitazones with
insulin have appeared; avoid such combinations. They should not be used during
pregnancy. The Diabetes Prevention Programme (2005) has shown that glitazones
have the potential to prevent type 2 DM in prediabetics.
GLUCOSIDASE INHIBITORS
Acarbose
It
is a complex oligosaccharide which reversibly inhibits αglucosidases, the
final enzymes for the digestion of carbohydrates in the brush border of small
intestine mucosa. It slows down and decreases digestion and absorption of
polysaccharides and sucrose: postprandial glycaemia is reduced without
increasing insulin levels. Regular use tends to lower Hb A1c, body
weight and serum triglyceride. These beneficial effects, though modest, have
been confirmed in several studies. Further, the stopNIDDM trial (2002) has
shown that longterm acarbose treatment in prediabetics reduces occurrence of
type 2 DM as well as hypertension and cardiac disease. In diabetics, it reduces
cardiovascular events.
Acarbose
is a mild antihyperglycaemic and not a hypoglycaemic; may be used as an
adjuvant to diet (with or without a sulfonylurea) in obese diabetics. Dose
50–100 mg TDS is taken at the beginning of each major meal. It is minimally
absorbed, but produces flatulence, abdominal discomfort and loose stool in
about 50% patients due to fermentation of unabsorbed carbohydrates.
GLUCOBAY
50, 100 mg tabs, ASUCROSE, GLUCAR 50 mg tabs.
Miglitol is similar to acarbose, and is more potent in inhibiting sucrase.
Related Topics
