Particulate carrier systems
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Drug delivery systems
Many particulate carriers have been designed for drug delivery and targeting. These include liposomes, micelles, microspheres, and nanoparticles.
Particulate
carrier systems
Many
particulate carriers have been designed for drug delivery and target-ing. These
include liposomes, micelles, microspheres, and nanoparticles. In general,
particulate carriers are phagocytosed by the macrophages of the mononuclear
phagocyte system (MPS), thereby localizing predominantly in the liver and
spleen. However, sterically stabilized particulate carriers have extended
circulation times. The in vivo fate
of particulate DDSs depends on the size, shape, charge, and surface
hydrophobicity of the particles.
Liposomes
Liposomes
are microscopic phospholipid vesicles. The phospholipid usually has a
hydrophilic headgroup and two hydrophobic chains. Phosphatidylcholine (PC), a
neutral phospholipid, has emerged as the major component used in the
preparation of liposomes. Phosphatidylglycerol and phosphatidylethanolamine are
also widely used. These lipid moieties spontaneously orient in water to give
the hydrophilic headgroup facing out into the aqueous environment and the lipid
chains orienting inward, thus avoiding the water phase; this gives rise to
bilayer structures. In general, liposomes can be multilamellar vesicles (MLVs),
which have diameters in the range of 1–5 μm. Extrusion or sonication of MLVs
results in the pro-duction of small unilamellar vesicles (SUVs) with diameters
in the range of 0.02–0.08 μm.
Large unilamellar vesicles (LUVs) can also be made by evaporation under reduced
pressure, resulting in liposomes with a diameter of 0.1–1 μm. The bilayer-forming lipid is the
essential part of the lamellar structure, while the other compounds are added
to impart certain charac-teristics to the vesicles.
Location
of a drug entrapped in the liposomes depends on the drug’s sol-ubility
characteristics. Water-soluble drugs can be entrapped in liposomes by
intercalation in the aqueous bilayers, while lipid-soluble drugs can be
entrapped within the hydrocarbon interiors of the lipid bilayers. Liposomes can
encapsulate small molecular weight drugs, proteins, peptides, oligonu-cleotides,
and genes. Examples of applications where liposomes have been successfully
employed to provide therapeutic benefit include amphotericin B liposomes. The
use of the antifungal agent amphotericin B formulated in liposomes has been
approved by the Food and Drug Administration (FDA) for treating systemic
mycoses.
The
rigidity and permeability of the bilayer strongly depend on the type and
quality of lipids used. The phospholipids bilayer can be two physi-cal states
based on their structural rigidity: gel state or a more fluid state known as
the liquid crystalline state.
Preference for either state depends on various characteristics of lipid
components, including (1) the chain length of hydrophobic alkyl chain, (2) the
degree of unsaturation of alkyl chain, and (3) the polar headgroup structure.
For example, a C18 saturated alkyl chain produces rigid bilayers
with low permeability at room temperature. The presence of cholesterol also
tends to rigidify the bilayers. This state also depends on temperature. Thus,
liposomes of a given lipid will assume gel state at a lower temperature, while
they become liquid crystalline at a higher temperature.
1. Types of liposomes
Liposomes
can be classified into the following categories: conventional liposomes,
stealth liposomes, targeted liposomes, and cationic liposomes.
1.1 Conventional liposomes
These
are neutral or negatively charged liposomes typically composed of only
phospholipids, glycolipids, and/or cholesterol, without derivatization, to
increase the circulation time. These liposomes are generally used for pas-sive
targeting to the phagocytic cells of the MPS, localizing predominantly in the
liver and spleen. Conventional liposomes have also been used for antigen
delivery.
1.2 Sterically stabilized (stealth) liposomes
Since
conventional liposomes are recognized by the immune systems as foreign bodies,
PEG–lipid (commonly known as PEGylated lipid), such as
PEG–phosphatidylethanolamine (PEG–PE) is often included in the prepa-ration of
liposomes. The PEGylated lipid reduces the uptake of liposomes by MPS or the
RES, resulting in prolonged circulation half-life. Simply persist-ing in the
bloodstream, PEGylated liposomes can localize into tumors and most sites of
inflammation. For an example, ALZA Corporation developed STEALTHTM liposomes,
which evade recognition by the immune system because of their unique PEG
coating. DoxilTM is a STEALTHTM liposome formulation of doxorubicin used for
the treatment of AIDS-related Kaposi’s sarcoma.
1.3 Targeted liposomes
In
addition to a PEG coating, most stealth liposomes also have some sort of
biological species attached as a ligand to the liposome to enable binding via a
specific expression on the targeted drug delivery site. These target-ing
ligands could be monoclonal antibodies (making an immunoliposome), vitamins, or specific antigens. Targeted liposomes
can target nearly any cell type in the body and deliver drugs that would
naturally be systemically delivered. Naturally toxic drugs can be much less
toxic if delivered only to diseased tissues.
1.4 Cationic liposomes
Cationic
liposomes are positively charged liposomes and used for nucleic acid delivery.
Cationic liposomes interact with the negatively charged phos-phate backbone of
DNA or RNA, leading to neutralization of the charge. Cationic liposomes are
prepared using a cationic lipid and a colipid, such as
dioleoylphosphatidylethanolamine (DOPE) or cholesterol. The cationic
amphiphiles differ markedly and may contain single multiple charges (primary,
secondary, tertiary, or quaternary). The three basic components of cationic
lipids include (1) a hydrophobic lipid anchor group, which helps in forming
liposomes and can interact with cell membranes; (2) a linker group; and (3) a
positively charged headgroup, which interacts with nucleic acids, leading to
nucleic acid condensation and charge neutralization. The linker group is an
important component that determines the chemical sta-bility and
biodegradability of the lipid. The physicochemical properties of
liposome/nucleic acid complexes are strongly influenced by the relative
pro-portions of each component and the structure of the headgroup.
2. Fabrication of liposomes
All
methods of making liposomes involve three to four basic stages: dry-ing down of
lipids from organic solvents (usually chloroform), dispersion of the lipid
mixtures in aqueous media, purification of the resultant lipo-somes, and
analysis of the final products. Figure 14.5
illustrates the stages common to different liposome preparation methods. The
drying down of large volume of organic solutions is most easily carried out in
rotary evaporator, fitted with a cooling coil and water bath. Rapid evaporation
of solvents is carried out by gentle warming (20°C–40°C) under pressure
(400–700 mmHg). The main difference between the various methods of preparation
is in the way in which the membrane components are dispersed in aqueous media.
The following methods are often used for dispersion of lipid membrane
components on hydration and agitation: extrusion, mechanical dispersion, microfluidization,
sonification, detergent dialysis, and ethanol injection. Hydrated lipid
solutions will initially form large, MLVs. After the initial pass through an
extrusion membrane, the par-ticle size distribution will tend toward a bimodal
distribution. After suf-ficient passes through the membrane, a unimodal, normal
distribution is
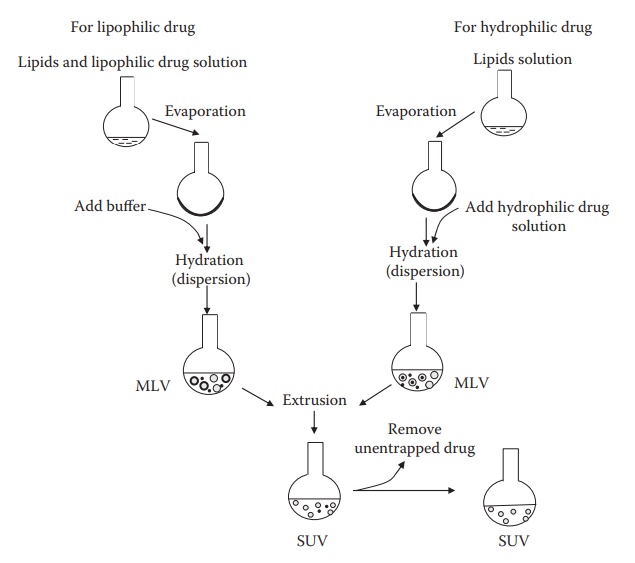
Figure 14.5 Stages common to different liposome preparation methods.
Microfluidizer is also used for
preparation of liposomes from concen-trated lipid suspensions. The
microfluidizer is a machine that pumps fluid at very high pressure (10000 psi,
which is 600–700 bar) through a 5-μm filter, after which it is forced
along microchannels, which then direct the two streams of fluid to collide
together at right angles at a very high veloc-ity. The fluid collected can be
recycled through the pump and interaction chamber until vesicles of the required
dimensions are obtained.
In
the ethanol injection method of liposome
preparation, an ethanol solution of lipids is injected rapidly into an
excess of saline or other aque-ous medium, through a fine needle. This
procedure can yield a high pro-portion of SUV of 25–50 nm; however, lipid
aggregates and larger vesicles may form if the mixing is not thorough enough.
The major limitations of ethanol injection method of liposome preparation are
(1) poor solubility of lipids in ethanol (40 mM for PC), (2) volume of ethanol
that can be introduced into the medium (7.5% v/v maximum), (3) poor drug
encapsula-tion efficiency, and (4) difficulty in removal of ethanol from
phospholipid membranes.
Microparticles and nanoparticles
A
microcapsule has drug located centrally
within the particle, where it is encased within a unique polymeric membrane.
The core can be solid, liq-uid, or gas, and the envelope is made of a
continuous, porous or nonporous polymeric phase. A drug can be dispersed inside
the polymeric envelope as solid particulates or dissolved in solution,
emulsion, suspension, or com-bination of both emulsion and suspension. In
contrast, a microsphere has its drug
dispersed throughout the particle; that is, the internal structure is a matrix
of drug and polymeric excipient (Figure 14.6).
Small molec-ular weight drugs, proteins, oligonucleotides, and genes can be
encapsu-lated into microparticles to provide their sustained release at disease
sites. A microcapsule is a
reservoir-type system in which drug is located centrally within the particle,
whereas a microsphere is a
matrix-type system in which drug is dispersed throughout the particle.
Microcapsules usually release their drug at a constant rate (zero-order
release), whereas microspheres typically give a first-order release of drugs.
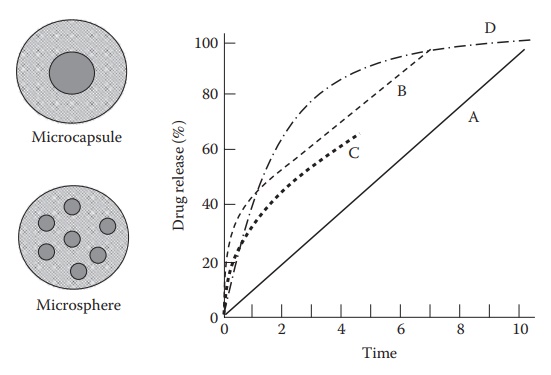
Figure 14.6 Schematic representation and drug- release profiles of microspheres and microcapsules: (a) microcapsules free of burst effect, (b) microcapsules with burst effect, (c) microspheres with square-root time release, and (d) micro-spheres with first-order release.
Fabrication of microparticulates
Microencapsulation is a technique that involves the encapsulation of small particles or solution of drugs in a polymer film or coat. Different methods of microencapsulation result in either microcapsules or microspheres. The most common methods of preparing microparticles and nanoparticles are emulsion and interfacial polymerization, and coacervation.
Emulsification
The
first step in almost any microencapsulation technique involves the for-mation
of an emulsion, usually of a polymeric solution inside a continuous phase.
Similarly, to disperse nonsoluble drugs inside polymeric solution, emulsions
must be created. Thus, a thorough understanding of emulsion formation and
properties is extremely important. The emulsion formation determines the
resulting particle size in the final process of encapsulation. An emulsion is
achieved by applying mechanical force, which deforms the interface between the
two phases to such an extent that droplets form. These droplets are typically
large and are subsequently disrupted or broken into smaller ones. The ability
to disrupt the larger droplets is a critical step in emulsification and in
encapsulation, where an emulsion is prepared. The size of the oil-phase
droplets obtained is determined by how rapidly the system is agitated when the
oil phase is added to the aqueous phase; it also determines the size of the
microparticles produced.
However,
protein and nucleic acid drugs are fairly labile and can be destroyed due to
the application of mechanical shear, and thus, preventive measures should be
taken to stabilize these drugs during emulsification process. A suitable
surfactant is needed to produce a stable emulsion, a result achieved by
lowering the surface tension. Devices commonly used for production of emulsions
are the following:
·
Ultrasonicator
·
Homogenizer
·
Microfluidizer
·
Injection
·
Stirring
·
Many more
Albumin
and some other water-soluble proteins can be used to prepare microspheres,
involving the formation of a water-and-oil (w/o) emulsion and stabilization of
the protein by cross-linking, using glutaraldehyde or heat denaturation. A
mixture of petroleum ether and cottonseed oil (60:40) containing 0.5% v/v Span®
80 can be used as a continuous phase
(~100 mL). Serum albumin is dissolved in phosphate-buffered saline (PBS)
con-taining 0.1% w/v sodium dodecyl sulfate (~1 mL). The albumin solution is
added dropwise to the continuous phase, stirred with 2,500 rpm with a
homogenizer. After 1 hour of mixing, glutaraldehyde
solution (100 μL, 5%–12%) is added
dropwise to the w/o emulsion, which is stirred for 1 hour at room temperature
to allow cross-linking. Alternatively, microspheres can be stabilized by heat denaturation at 100°C–120°C.
Following stabiliza-tion, microspheres are freed of oil by washing with
petroleum ether (x3) and isopropanol (x2); they are then suspended in PBS and
stored at 4°C until required. Biodegradation of albumin microspheres and
drug-release rate are dependent on the concentration of glutaraldehyde
concentration or degree of heat denaturation. Apart from albumin, other
proteins such as hyal-uronidase and
chitosan can also be used for preparation of microspheres by using cross-linkers.
Solvent evaporation
Solvent
evaporation is the most popular method of preparation of mic-roparticles. A
core material and capsule wall material are dissolved in a water-immiscible, volatile organic solvent, and the resulting
solution is emulsified in an aqueous
solution. The solvent is allowed to evaporate, thereby producing solid microcapsules
or microparticles. Another way is to form a double emulsion, where an aqueous
core material solution is emulsi-fied in a polymer-volatile organic solvent
solution. The resulting emulsion is emulsified in water, giving a double
emulsion. Evaporation of the volatile solvent yields a solid microcapsule with
an aqueous core. Methylene chloride (CH2Cl2) is a preferred solvent because of its volatility
(boiling point, 41°C) and its capacity of dissolving broad range of polymers.
Chloroform and ethyl acetate can also be used. A mixture of methylene chloride
(a water-immiscible solvent) and acetone (a water-miscible solvent) can also be
used.
The
added drug may be completely dissolved in the polymer solution, or it may be
completely insoluble and simply form a dispersion, suspension, or
suspension–emulsion. In the latter case, the solid particles must be
micron-ized, so that their mean diameter is much less than the desired mean
micro-sphere size. To aid emulsification, a surfactant is normally dissolved in
the water phase before the oil-in-water emulsion is formed. A good example is
partially hydrolyzed (88%) poly(vinyl alcohol) (PVA).
After
obtaining desired droplet size and emulsion stability, the system is stirred at
constant rate, followed by solvent evaporation by using a rotary evaporator.
Following solvent evaporation, the microparticles are separated from the
suspending medium by filtration or centrifugation, washed, and dried. The
maximum drying temperature must remain below the glass temperature of the
polymer encapsulant or the microspheres fuse together. Although the solvent
evaporation process is conceptually simple, the nature of the product can be
affected by the following factors:
·
Polymer molecular weight and concentration
·
Polymer crystallization
·
Type of drug and method of incorporation (solid, liquid, and
suspension)
·
Organic solvent used
·
Type and concentration of surfactant used in the aqueous
phase
·
Ratio of organic phase to aqueous phase
·
Rate of stirring
·
Evaporation temperature
In
general, semicrystalline polymers often give porous structures, with
spherulites on the surface of the microspheres. Uniform, pore-free spheres are
most readily obtained with amorphous polymers.
Biodegradable
polylactide (PLA) and its copolymer with glycolide (polylactide-co-glycolide
[PLGA]) are commonly used for preparing mic-roparticles, from which the drug
can be released slowly over a period of a month or so. Microspheres can be used
in a wide variety of dos-age forms, including tablets, capsules, and suspensions.
Table 14.1 lists some of the FDA-approved commercial products of microspheres. Lupron Depot from TAP Pharmaceuticals is
an FDA-approved prepa-ration of PLGA microspheres for sustained release of a
small-peptide luteinizing hormone-releasing hormone (LHRH) agonist. More
recently, PLGA microspheres of recombinant human growth hormone have been
developed and marked successfully by Genentech, Inc., under the trade name of
Nutropin Depot. Polylactide-co-glycolide degrades into lactic and glycolic
acids.
Interfacial (or in situ) polymerization
In
interfacial polymerization,
oil-soluble monomers and water-soluble monomers react at the water/oil
interface of w/o or o/w dispersions, result-ing in the formation of polymeric
microcapsules. The process involves an initial emulsification step, in which an
aqueous phase, containing a reactive monomer and a core material, is dispersed
in a nonaqueous phase. This is followed by the addition of a second monomer to
the continuous phase. Monomers in the two phases then diffuse and polymerize at
the interface to form a thin film. The most widely used example of microcapsule
preparation using this method is the interfacial polymerization of
water-soluble alkyl-diamines with oil-soluble acid dichlorides to form
polyamides. Examples of other polymeric wall materials include polyurethanes,
polysulfonamides, polyphthalamides, and poly(phenyl esters). Interfacial
polymerization of a monomer almost always produces microcapsules, whereas
solvent evapora-tion may result in microspheres or microcapsules, depending on
the amount of drug loading.
Complex coacervation
Complex
coacervation uses the interaction of two oppositely charged poly-electrolytes
in water to form a polymer-rich coating solution called a com-plex coacervate.
This solution (or coacervate) engulfs the liquid or solid being encapsulated,
thereby forming an embryo capsule. Cooling the sys-tem causes the coacervate
(or coating solution) to gel via network forma-tion. Gelatin and gum arabic are
primary components of most complex coacervation systems. Coacervation uses the
common phenomenon of polymer–polymer
incompatibility to form microcapsules. The first step is to form a solution of gelatin in
deionized water at 11 wt% and 45°C–55°C. Once the gelatin and gum arabic
solutions are prepared, the drug is emulsi-fied or dispersed in the 45°C–55°C
gelatin solution. Once the drug–gelatin emulsion or dispersion is formed, it is
diluted by addition of a known volume of the 45°C–55°C deionized water and
11wt% gum arabic solution (45°C–55°C). The pH of the resulting mixture is
adjusted to 3.8–4.4 by addition of acetic acid. After the pH is adjusted, the
system is allowed to cool down to room temperature and then to below 10°C, and
at this point, glutaraldehyde is slowly added to cross-link the polymer. The
system is stirred gently throughout this cooling period.
Alginates form gels
on reaction with calcium salts: These gels consist of almost 99% of water and 1% or less
of alginate. Cross-links are caused either by simple ionic bridging of two
carboxyl groups on adjacent poly-mer chains via calcium ions or by chelation of
single calcium ions by hydroxyl and carboxyl groups on each of the pair of
polymer chains. Several types of viable cells (erythrocytes, sperm cells,
hepatoma cells, and hepatocytes), tissues (pancreatic endocrine tissues and
islets), and other labile biological substances are encapsulated within
semipermeable alginate microspheres. The process involves suspending the living
cells or tissues in sodium alginate solution, and the suspension is then
extruded to produce microdroplets, which fall into a CaCl2 solution
and form gelled microbeads with the cells or tissues entrapped. These
microbeads are next treated with polylysine solution, which displaces the
surface layer of calcium ions and forms a permanent polysalt shell or membrane.
Porous
microspheres are formed by gelation of the following:
·
Sodium alginate and chitosan
·
Sodium alginate and CaCl2
·
Sodium alginate and polylysine
Hot melt microencapsulation
In
hot melt microencapsulation, melted polymers are mixed with drugs, and the
mixture is then suspended in an immiscible solvent that is heated at 5°C above
the melting point of the polymer and stirred continuously. Once the emulsion is
stabilized, it is cooled until the core material has solidified. The solvents
used in this process are usually silicon and olive oils. After cooling,
microspheres are washed with petroleum ether to have free-flow powders. To
avoid degradation of drugs due to heat, polymers with low melting points are
used in this process. Robert Langer and his associates used this method to
prepare polyanhydride microspheres.
Solvent removal
In
the solvent-removal process, the fabrication occurs at the room temper-ature
totally in organic solvents, which is good for hydrolytically labile polymers
such as polyanhydrides and water-soluble drugs. Mathiowitz and Langer used this
method to encapsulate zinc insulin into polyanhy-dride, such as
poly(carboxyphenoxypropane-co-sebacic acid) (poly(CPP-SA), 50:50, microspheres.
In this example, the polymer was dissolved in methylene chloride, the desired
amount of the drug was added, and then the mixture was suspended in silicon oil
containing Span 85. Petroleum ether was then added, and the mixture was stirred
until methylene chloride was extracted into the oil solution and sufficient
microcap-sule hardening was achieved. The microspheres were isolated by
filtra-tion, washed with petroleum ether, and dried overnight under vacuum. The
solvent-removal process is somewhat different from organic-phase separation (or
coacervation) process. In solvent removal, the polymeric solution is introduced
into the continuous phase, an emulsion is formed first, and then the organic
solvent is extracted into the continuous phase. In the organic-phase
separation, the polymer is dissolved in the continu-ous phase, a phase inducer
is introduced, a coacervate is formed, and, finally, drug encapsulation occurs.
Spray drying
In
this process, polymers are dissolved in a volatile solvent, such as meth-ylene
chloride. The spray-drying process involves dispersion of the core material in
a solution of coating substance and spraying the mixture into an environment
that causes the solvent to evaporate. This method is often used to encapsulate
heat-sensitive drugs in polyanhydride microspheres.
Nanoparticles
Nanoparticles
are solid colloidal particles ranging in size from 10 to 1,000 nm. Depending on
the fabrication process, two different types of nanoparticles can be obtained,
namely nanospheres and nanocapsules. Nanospheres have a matrix-type structure,
in which a drug is dispersed, whereas nanocap-sules exhibit a membrane-wall
structure, with an oily phase containing the drug. Because these nanoparticles
have very high surface areas, drugs may also be adsorbed on their surface.
Biodegradable nanoparticles from poly(lactic acid)-poly(glycolic acid) (PLGA),
polycaprolactone, and poly-alkylcyanoacrylates have been widely studied.
Gelatin nanoparticles are also used, which are prepared by desolvation of a
gelatin solution con-taining drug bound to the gelatin. Hardening of the
gelatin nanoparticles is achieved by glutaraldehyde, which cross-links with
gelatin and is more efficient than formaldehyde.
Related Topics
