Pragmatic Solutions
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Periodic Safety Update Reports
The PSUR process relies heavily on the availability of adequate resources, particularly since CIOMS V intro-duced the concept of PSURs covering periods longer than 6 months .
PRAGMATIC SOLUTIONS
In
a recent paper, Michael J. Klepper of North Carolina-based Integrated Safety Systems,
Inc., a safety surveillance and consulting firm for phar-maceutical, biological
and medical device compa-nies, outlined some of the ways that companies could
maximise the efficiency of their procedures for producing PSURs, avoid
potential pitfalls and ensure full compliance (Klepper, 2004):
RESOURCE PLANNING
The
PSUR process relies heavily on the availability of adequate resources,
particularly since CIOMS V intro-duced the concept of PSURs covering periods
longer than 6 months (including the five yearly reports for local product
renewals in Europe) which still have to be submitted within 60 days of the DLP.
The resources needed depend on factors including: the size of the company, the
number of marketed products, when these products were approved, the number of
coun-tries where these medical products are marketed, the volume of ADRs and
the complexity of the medi-cal condition for which the medical product is
indi-cated. For example, the process of producing a PSUR for a newly approved
AIDS drug that is marketed in many countries will require considerably more
resources than the same process for a 15-year-old topical formulation, which is
only approved in a few countries worldwide for the treatment of athlete’s foot.
Those resources are not solely restricted to the product safety department. As
in the Lundbeck SOP, contributions are also required from regulatory
departments, which provide information regarding the status of worldwide
approval and any regulatory action taken anywhere in the world; clinical
research departments, which provide data on any important safety issues
emerging from ongoing clinical trials and marketing/financial services
departments, which hold the sales/prescriptions data needed to estimate patient
exposure. Summary bridging reports and addendum reports require additional
resources. When allocating resources to the PSUR process, companies should also
be aware that the same departments will be called upon to produce the clinical
trial annual reports required under the EU Clinical Trials Directive (see Clinical Trial Annual Report). Over a
given period, say a year, the MAH
should know the number of PSURs due in that year, including the DLPs and
submission dates of these reports. It should also factor in an estimate of
volume and complexity of cases. The MAH can then allocate its resources
accordingly and put in place a contingency plan in case new work arises, for
example an unexpected regulatory query. If there are too few resources
available, the MAH may consider outsourcing the work, hiring more people,
providing more training or re-prioritising projects. It is also essential that
communication between depart-ments is good, so that all the personnel involved
in producing the PSUR are aware of expectations, deliv-erables and dates of
completion.
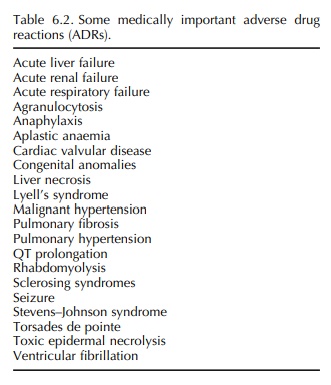
DEFINITIONS AND SCRIPTS FOR MEDICALLY IMPORTANT ADRS
Reported
ADR data are, in general, incomplete and of poor quality (Venulet, 1986).
Although most suspected ADRs are reported by physicians trained in what is
called Western medicine, there are consid-erable cultural differences in the
use and interpre-tation of certain medical terms. Reporting Adverse Drug
Reactions: Definitions of Terms and Crite-ria for Their Use (CIOMS, 1999)
is one attempt to cross those
cultural differences by establishing standard definitions for selected terms
for ADRs and minimum requirements for the use of those terms in international
reporting. In an introductory chapter to that book, Ronald Mann, former
direc-tor of the University of Southampton’s Drug Safety Research Unit,
emphasises the importance of keeping the patient’s own words when reporting
complaints, so as not to corrupt the data at source. At the next stage of the
communication process – when the physician-reporter passes the information on
to a company representative – Klepper suggests that scripts should be developed
that are designed to extract the criti-cal information from the reporter. Those
responsible for the intake of ADR information should be thor-oughly trained in
the use of these scripts. A script dealing with liver necrosis, for example,
would guide the representative to ask specific questions, such as the basis of
the diagnosis (e.g. viral serologies and needle biopsy). Examples of some
medically impor-tant ADRs (FDA, 2003; Mann, 2005) are summarised in Table 6.2.
The World Health Organization Critical Term list provides an even more
extensive list of such ADRs (WHO, 1998).
TRAINING
The
personnel involved in the PSUR process require training in four broad areas:
·
Product training: To fully
understand a product’s pharmacology
or biological activity, mechanism of action and the known risks associated with
its use;
·
Clinical training: To fully
understand the charac-teristics of the targeted patient population likely to
take the product, with respect to underlying comor-bidities and concomitant
medications;
·
Pharmacovigilance
training:
To fully understand the critical
concepts, disciplines, and components associated with pharmacovigilance, the
methods used with key considerations affecting risk versus benefits analysis
and the medical significance of the most important ADRs and
·
MedDRA training: To fully
understand the dictionary, its hierarchy and the implications of its
granularity (see ‘STANDARDISED AND HARMONISED MEDDRA CODING’).
STANDARDISED AND HARMONISED MEDDRA CODING
One
of the characteristics of MedDRA that distin-guishes it from traditional
dictionaries is its extreme specificity or granularity. Slightly different
verba-tim terms are prone to be coded to different preferred terms and even
entirely different SOCs, with important implications for subsequent
statisti-cal analyses. The quality of the term used by the reporter (verbatim
term) drives the coding process. A high quality verbatim term is likely to
autoencode, whereas a poor quality term is more likely to require manual
assignment of a MedDRA term, which in turn increases the potential for
inconsistencies. To ensure coding consistency for global companies where it is
likely that cases will be entered remotely into the MAH’s central database, a
global coding convention should be created, maintained and revised as
neces-sary. This document could include, for example, the Points to consider developed by the Maintenance and Support Services Organization for
MedDRA (MSSO, 2006), as well as other conventions. An example of a coding
convention would be the establishment of a ‘rule’ that states that for any
surgical procedure, the ADR that led to the surgery will be coded rather than the
procedure itself, e.g. ‘gallstones’ rather than ‘cholecystectomy’.
PRESPECIFIED SEARCH CRITERIA
Prespecified search criteria for data retrieval should be
developed, used and documented. This will ensure consistent and reproducible
data retrieval.
ONGOING MEDICAL REVIEW
Because
the presentation of individual case histories and the overall safety evaluation
are the most time-consuming parts of the PSUR process, companies should commit
themselves to an ongoing review process, regardless of when a PSUR falls due.
It is also advisable to set up an in-house safety review commit-tee, as
Lundbeck has done. The medical reviewer responsible for a given medical product
may become too close to the data to judge it objectively and may end up
overlooking signals. The safety review committee should be composed of senior,
experienced individuals who are not directly involved in the safety evaluation
of the medical product. This committee should meet regularly, say quarterly, to
take a fresh look at the data and to bring to the review process a broader
medical expertise than was available in the initial evaluation.
METRICS
Measures should be put in place to monitor existing
processes, to ensure that they remain effective and efficient and that
corrective actions are having the intended effect. An example of such a metric
would be looking at the number of avoidable ADRs that were due to a newly
identified drug–drug interaction. Risk management initiatives could be put in
place to address such a finding, such as a label change or patient education.
The results of these initiatives should be reflected in subsequent PSURs. Other
examples of PSUR metrics are summarised in Table 6.3.

Related Topics
