PSUR Content
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Periodic Safety Update Reports
PSURs contain proprietary information, so the title page should contain a statement on the confidentiality of the data and conclusions included in the report.
PSUR CONTENT
The
amendment to ICH E2C stipulates that the MAH should submit a PSUR to the
competent authority of the country or region in question with succinct summary
information and a benefit–risk analysis in the light of new or changing
post-authorisation infor-mation. Specifically, the contents of the PSUR should
be as laid out in Table 6.1. The rest of this section describes an overview of
a model PSUR.
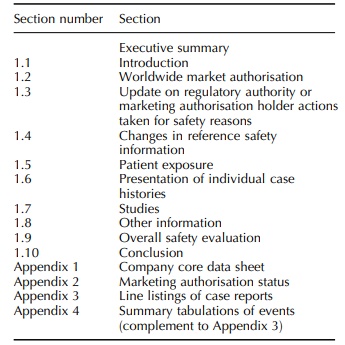
TITLE
PSURs
contain proprietary information, so the title
page should contain a statement on
the confidentiality of the data and
conclusions included in the report.
EXECUTIVE SUMMARY
The
executive summary should consist of a
brief overview providing the reader with a description of the most important
information. An example can be found on page 333 of the CIOMS V report (CIOMS,
2001).
INTRODUCTION
The
introduction sets the scene and puts
the report in context, cross-referencing it to previous reports, describing
those products/formulations that are included and excluded, outlining the
pharmacology of the product, its indications (both marketed and in clinical
trials) and any co-licensing agreements.
WORLDWIDE MARKETING AUTHORISATION STATUS
The PSUR should include a short summary of the worldwide marketing authorisation status and cross-reference this to an appendix in which the cumulative approvals (and renewal dates) should be tabulated in chronological sequence. This table should also include lack of approval, relevant explanations from regula-tory authorities and withdrawals by the company for efficacy or safety reasons.
UPDATE ON REGULATORY AUTHORITY OR MAH ACTIONS TAKEN FOR SAFETY REASONS
The
update on regulatory authority or MAH
actions taken for safety reasons refers
to
• marketing authorisation, withdrawal or suspension;
• failure to obtain a marketing authorisation renewal;
• restrictions on distribution;
• clinical trial suspension;
• dosage modification/formulation changes and
• changes in target population or indications.
The
update should discuss the safety-related reasons that led to the actions
described and append the appropriate documentation including any communica-tion
with healthcare professionals (e.g. ‘Dear Doctor’ letters).
CHANGES IN REFERENCE SAFETY INFORMATION
The
changes in reference safety information
section refers to changes in the CCSI. The CCDS, which incorporates the CCSI,
should be included as an appendix. If no CCDS is available, a national SPC can
be used. A covering letter should discuss meaningful differences between the
CCSI and local datasheets and comment on the consequences for safety
evaluations and for actions proposed or initiated.
PATIENT EXPOSURE
Patient exposure refers to both
market exposure and clinical trials
(if relevant). Estimates of patient expo-sure for marketed drugs often rely on
gross approxi-mations of in-house or purchased sales data or volume. This
information is not always reliable or available for all products. For example,
hospital-based statistics from the major use-monitoring sources are frequently
unavailable. It is also difficult to obtain accurate data for generics,
non-prescription drugs or multiple drug regimens. The MAH should use a
consistent method of calculation across PSURs for the same product. If a change
in the method is appropriate, both previ-ous and current methods and
calculations should be shown in the PSUR introducing the change. When exposure
data are based on information from a period that does not fully cover the
period of the PSUR, the MAH can make extrapolations using the available data.
When this is done, it should be clearly indicated what data were used and why
it is valid to extrapolate for the PSUR period in question (for example stable
sales over a long period and seasonality of use of the product). The CIOMS V
report contains examples of patient exposure estimations (CIOMS, 2001).
PRESENTATION OF INDIVIDUAL CASE HISTORIES
There
is no specific guidance in E2C on the presentation
of individual case histories, but because it is impractical to present all case reports for the reporting
period, a brief description of the crite-ria used to select cases for
presentation should be given. This section of the PSUR should contain a
description and analysis of selected cases, including fatalities, presenting
new and relevant safety infor-mation and grouped by medically relevant
head-ings or system organ classes (SOCs). Depending on their type or source,
available ADR cases should be presented as line listings and/or as summary
tabu-lations. A line listing provides key information but not necessarily all
the details customarily collected on individual cases. However, it does serve
to help regulatory authorities identify cases which they might wish to examine
more closely by requesting full case reports. In addition to individual case
line listings, summary tabulations of ADR terms for signs, symp-toms and
diagnoses across all patients should usually be presented. Such tabulations
should be based on the data in line listings (e.g. all serious ADRs and all
non-serious unlisted ADRs) but also on other sources for which line listings
are not requested (e.g. non-serious listed ADRs).
STUDIES
Studies refer to only those
company-sponsored studies and
published safety studies, including epidemiology studies, that produce findings
with potential impact on product safety information. These should be included
along with a discussion of any final or interim results. The MAH should not
routinely catalogue or describe all the studies.
OTHER INFORMATION
Other information may include risk
management programmes the MAH has put
in place and/or a benefit–risk analysis report. If such an analysis has been
conducted separately, a summary of the anal-ysis should be included in this
section. This section can also include important information received after the
DLP, e.g. significant follow-up on cases included in the PSUR and changes to
the CCSI agreed after the DLP.
OVERALL SAFETY EVALUATION
The
overall safety evaluation should
highlight new information on serious and non-serious unlisted ADRs. For listed
ADRs, it should describe any reported changes in the characteristics of the
reaction (e.g. severity, outcome and target population) as well as increases in
frequency of reporting of reactions. For emerging safety issues, the
information received during the period under review should be discussed from
the perspective of cumulative experience. For new safety issues, the current
action should be stated (e.g. under active review). If there are no new safety
issues, this should be stated with a note that the information is in keeping
with the established safety profile. All evaluations should be concise, and the
discussion and analysis should be organised by SOC rather than by listedness or
seriousness. Although related terms might be found in different SOCs, they
should be reviewed together for clinical relevance. This section should also
review reports of
·
drug interactions;
·
overdose: deliberate or accidental and treatment;
·
abuse or misuse;
·
pregnancy or lactation: positive and negative expe-riences;
·
special patient groups (e.g. children, elderly, organ
impaired) and
·
effects of long-term treatment.
CONCLUSION
The
conclusion should indicate safety
data which are not in accordance with previous experience and/or with the CCSI
and specify and justify any action recommended or initiated.
APPENDICES
Although the intent of the PSUR initiative is to have a standard PSUR format and content, individual coun-tries may require additional information. For example, the PSUR is designed to contain information reported or confirmed by a healthcare professional, but regula-tory agencies in some countries, including the US, also require consumer reports of ADRs. This is accom-modated by including consumer information in an Appendix to the PSUR.
Related Topics
