The Basis of Pharmacovigilance
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: The Basis of Pharmacovigilance
Pharmacovigilance – the study of the safety of marketed drugs under the practical conditions of clinical use in large communities – involves the para-dox that what is probably the most highly regulated industry in the world is, from time to time, forced to remove approved and licensed products from the market because of clinical toxicity.
THE BASIS OF PHARMACOVIGILANCE
Introduction
‘Not all hazards can be known before a drug is
marketed’.
Committee
on Safety of Drugs, Annual Report 1969, 1970.
Pharmacovigilance
– the study of the safety of marketed drugs under the practical conditions of
clinical use in large communities – involves the para-dox that what is
probably the most highly regulated industry in the world is, from time to time,
forced to remove approved and licensed products from the market because of
clinical toxicity. Why is such close regulation not effective in preventing the
withdrawal of licensed products? The question has been with us from the very
early days of the 1960s and remains with us today, and its consideration tells us
a great deal about pharmacovigilance.
The
greatest of all drug disasters was the thalido-mide tragedy of 1961–62.
Thalidomide had been introduced, and welcomed, as a safe and effective hypnotic
and anti-emetic. It rapidly became popular for the treatment of nausea and
vomiting in early pregnancy. Tragically, the drug proved to be a potent human
teratogen that caused major birth defects in an estimated 10 000 children in
the countries in which it was widely used in pregnant women. Figure 1.1 shows a
child with thalidomide-induced amelia of the upper limbs and phocomelia of the
lower limbs fitted with the kind of prostheses available at that time. The
story of this disaster has been reviewed elsewhere (Mann, 1984).
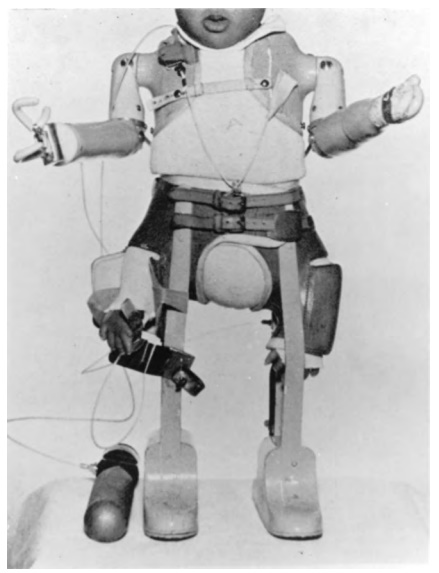
Figure 1.1. Child with thalidomide-induced deformities of the upper and lower limbs fitted with pneumatic prostheses.
The
thalidomide disaster led, in Europe and else-where, to the establishment of the
drug regulatory mechanisms of today. These mechanisms require that new drugs
shall be licensed by well-established regu-latory authorities before being
introduced into clini-cal use. This, it might be thought, would have made
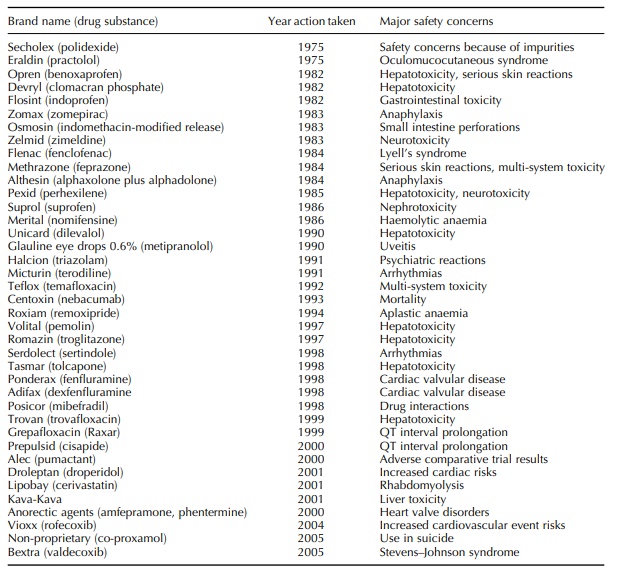
medicines safe – or, at least, acceptably safe. But Table 1.1 summarizes a list
of 39 licensed medicines withdrawn, after marketing, for drug safety reasons
since the mid-1970s in the United Kingdom.
Table 1.1. Drugs withdrawn in the United Kingdom by the marketing authorization holder or suspended or revoked by the Licensing Authority.
Why
should the highly regulated pharmaceutical industry need, or be compelled, to
withdraw licensed medicines for drug safety reasons? Why do these problems of
licensed products being found toxic continue despite the accumulated experience
of more than 45 years since the thalidomide tragedy?
Partly, the problem is one of numbers. For example, the median number of patients contributing data to the clinical safety section of new drug licensing applications in the United Kingdom is only just over 1500 (Rawlins and Jefferys, 1991). Increasing regulatory demands for additional information before approval have presumably increased the average numbers of patients in applications, especially for new chemical entities; nevertheless, the numbers remain far too small to detect uncommon or rare adverse drug reactions (ADRs), even if these are serious.
The
size of the licensing applications for impor-tant new drugs cannot be
materially increased without delaying the marketing of new drugs to an extent
damaging to diseased patients. Thus, because of this problem with numbers, drug
safety depends very largely on the surveillance of medicines once they have
been marketed.
A
second reason for difficulty is that the kinds of patients who receive licensed
medicines are very different from the kinds of volunteers and patients in whom
pre-marketing clinical trials are undertaken. The patients in formal clinical
trials almost always have only one disease being treated with one drug. The
drug, once licensed, is likely to be used in an older group of patients, many
of whom will have more than one disease and be treated by polypharmacy. The
drug may also be used in paediatric patients, who are generally excluded from
initial clinical trials. The formal clinical trials may be a better test of
efficacy than they are of safety under the practical conditions of everyday
clinical usage.
A
third problem is that doctors may be slow or inef-fective in detecting and
reporting adverse drug effects. Many of the drugs summarized in Table 1.1 were
in widespread, long-term use before adverse reactions were detected, and even
now, hospital admissions due to ADRs have shown an incidence of between 2.4%
and 3.6% of all admissions in Australia with simi-lar or greater figures in
France and the United States (Pouyanne et
al., 2000). Even physicians astute in detecting adverse drug effects are
unlikely to identify effects of delayed onset.
A
fourth reason for difficulty is that drugs are often withdrawn from the market
for what may be very rare adverse effects – too infrequent by far to have shown
up in the pre-licensing studies – and we do not yet have effective means in
place for monitoring total post-marketing safety experience. This situation may
well change as large comprehensive databases such as the General Practice
Research Database (GPRD) become more widely used for signal detection and
evaluation. These databases record, in quite large and representative
populations, all usage of many specific medicines and clinical outcomes and can
be used to systematically screen for and evaluate serious adverse events.
Because they contain comprehensive infor-mation on some important information,
such as age, sex, dose and clinical events on all patients in the represented
population, they are systematic compared with spontaneous reporting systems.
They may offer a better chance of detecting long-latency adverse reac-tions,
effects on growth and development and other such forms of adverse experience.
Some
of the difficulties due to numbers, patient populations and so on were
recognized quite early. The Committee on Safety of Drugs in the United Kingdom
(established after the thalidomide disas-ter, originally under the chairmanship
of Sir Derrick Dunlop, to consider drug safety whilst the Medicines Act of 1968
was being written) said – quite remarkably – in its last report (for 1969 and
1970) that ‘no drug which is pharmacologically effective is without hazard.
Furthermore, not all hazards can be known before a drug is marketed’. This then
has been known for over 35 years. Even so, many prescribers still seem to think
that licensed drugs are ‘safe’, and they are surprised when a very small
proportion of licensed drugs have to be withdrawn because of unex-pected drug
toxicity. Patients themselves may have expectations that licensed drugs are
‘completely safe’ rather than having a safety profile that is acceptably safe
in the context of the expected benefit and nature of the underlying health condition.
The
methodological problems have been long recognized. The Committee on Safety of
Medicines, the successor in the United Kingdom to the Dunlop Committee,
investigating this and related problems, established a Working Party on Adverse
Reactions. This group, under the chairmanship of Professor David Grahame-Smith,
published its second report in July 1985. The report supported the continuation
of methods of spontaneous reporting by professionals but recommended that
post-marketing surveillance (PMS) studies should be undertaken on
‘newly-marketed drugs intended for widespread long-term use’; the report also
mentioned record-linkage meth-ods and prescription-based methods of drug safety
surveillance as representing areas of possible progress (Mann, 1987).
Similar
reviews and conclusions have emerged from the United States since the
mid-1970s. A series of events in the United States recently created a
resurgence of interest in drug safety evaluation and management. The
Prescription Drug User Fee Act (PDUFA) of 1992 provided additional resources at
the Food and Drug Administration (FDA) for drug reviews through user fees and
established target time-lines for FDA reviews. The shorter approval times lead
to some medications being approved sooner in the United States than that in
Europe in contrast to the pre-PDUFA experience. A few highly visible drug
withdrawals led to a perception that perhaps drugs were being approved too
quickly. Lazarou, Pomeranz and Corey (1998) published the results of a
meta-analysis that estimated that 106 000 fatal adverse reac-tions occurred in
the United States in 1994. This and other articles (Wood, Stein and Woosley,
1998) stimulated considerable public, congressional and regulatory attention on
reducing the societal burden of drug reactions and medication errors (Institute
of Medicine, 1999; U.S. Food and Drug Administra-tion, 1999; United States
General Accounting Office, 2000). As a result, greater attention and resources
are currently being devoted to signal generation and eval-uation by the FDA,
industry and academic centres. Moreover, efforts are underway to develop better
tools to manage recognized risks through a variety of inter-ventions, such as
communications with healthcare providers and patients, restricted product
distribution systems and other mechanisms. Additional effort is being focused
on measuring the success of these risk-management interventions. This new
initiative repre-sents a fundamental shift in the safety paradigm in the
successor in the United Kingdom to the Dunlop Committee, investigating this and
related problems, established a Working Party on Adverse Reactions. This group,
under the chairmanship of Professor David Grahame-Smith, published its second
report in July 1985. The report supported the continuation of methods of
spontaneous reporting by professionals but recommended that post-marketing
surveillance (PMS) studies should be undertaken on ‘newly-marketed drugs
intended for widespread long-term use’; the report also mentioned
record-linkage meth-ods and prescription-based methods of drug safety
surveillance as representing areas of possible progress (Mann, 1987).
Similar
reviews and conclusions have emerged from the United States since the
mid-1970s. A series of events in the United States recently created a
resurgence of interest in drug safety evaluation and management. The
Prescription Drug User Fee Act (PDUFA) of 1992 provided additional resources at
the Food and Drug Administration (FDA) for drug reviews through user fees and
established target time-lines for FDA reviews. The shorter approval times lead
to some medications being approved sooner in the United States than that in
Europe in contrast to the pre-PDUFA experience. A few highly visible drug
withdrawals led to a perception that perhaps drugs were being approved too
quickly. Lazarou, Pomeranz and Corey (1998) published the results of a
meta-analysis that estimated that 106 000 fatal adverse reac-tions occurred in
the United States in 1994. This and other articles (Wood, Stein and Woosley, 1998)
stimulated considerable public, congressional and regulatory attention on
reducing the societal burden of drug reactions and medication errors (Institute
of Medicine, 1999; U.S. Food and Drug Administra-tion, 1999; United States
General Accounting Office, 2000). As a result, greater attention and resources
are currently being devoted to signal generation and eval-uation by the FDA,
industry and academic centres. Moreover, efforts are underway to develop better
tools to manage recognized risks through a variety of inter-ventions, such as
communications with healthcare providers and patients, restricted product
distribution systems and other mechanisms. Additional effort is being focused
on measuring the success of these risk-management interventions. This new
initiative repre-sents a fundamental shift in the safety paradigm in the United
States and offers new challenges to phar-macovigilance professionals. In fact,
the shift is not restricted to the United States as both the FDA and the EMEA
in 2005 issued guidance documents for industry on signal detection, evaluation,
good pharma-covigilance practice and recommendations for manag-ing risks after
the approval (EMEA, 2005; U.S. Food and Drug Administration, 2005a–c).
We
have long recognized then that the safety of patients depends not only on drug
licensing by regu-latory bodies but also on post-marketing drug safety
surveillance, pharmacovigilance. It is also important to note that the same
post-marketing information needed to confirm new safety signals is also needed
to refute signals and protect the ability of patients to benefit from needed
medicines that may be under suspicion due to spurious signals.
