The Genetic Basis of ADRS
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Pharmacogenetics and the Genetic Basis of ADRs
The CYP P450 monooxygenase system of enzymes detoxifies xeno-biotics and activates procarcinogens and promutagens in the body through oxidative metabolic pathways.
THE GENETIC BASIS OF ADRS
POLYMORPHISMS AFFECTING DRUG METABOLISM
Most drugs are degraded through a
limited number of metabolic pathways, most of which involve micro- somal
hepatic enzymes. Ingelman-Sundberg et al.
(1999) reported that about 40% of this human cytochrome (CYP) P450-dependent
drug metabolism is carried out by polymorphic enzymes capable of altering these
metabolic pathways. The CYP P450 monooxygenase system of enzymes
detoxifies xeno-biotics and activates procarcinogens and promutagens in the
body through oxidative metabolic pathways. These enzymes play an important role
in the elim-ination of endogenous substrates (such as choles-terol) and
lipophilic compounds [such as central nervous system (CNS) drugs that cross the
blood-brain barrier], which otherwise tend to accumulate to toxic
concentrations. This very large and well-studied gene family consists of many
isoforms – for example over 70 variant alleles of the CYP2D6 locus have been
described (Ingelman-Sundberg et al.,
1999). The distribution of variant alleles for these enzymes differs among
ethnic and racial subpopula-tions, with significant implications for clinical
practice in various areas (Table 51.1). Alleles causing altered (enhanced or
diminished) rates of drug metabolism have been described for many of the P450
enzymes, and the underlying molecular mechanisms have been identified for some.
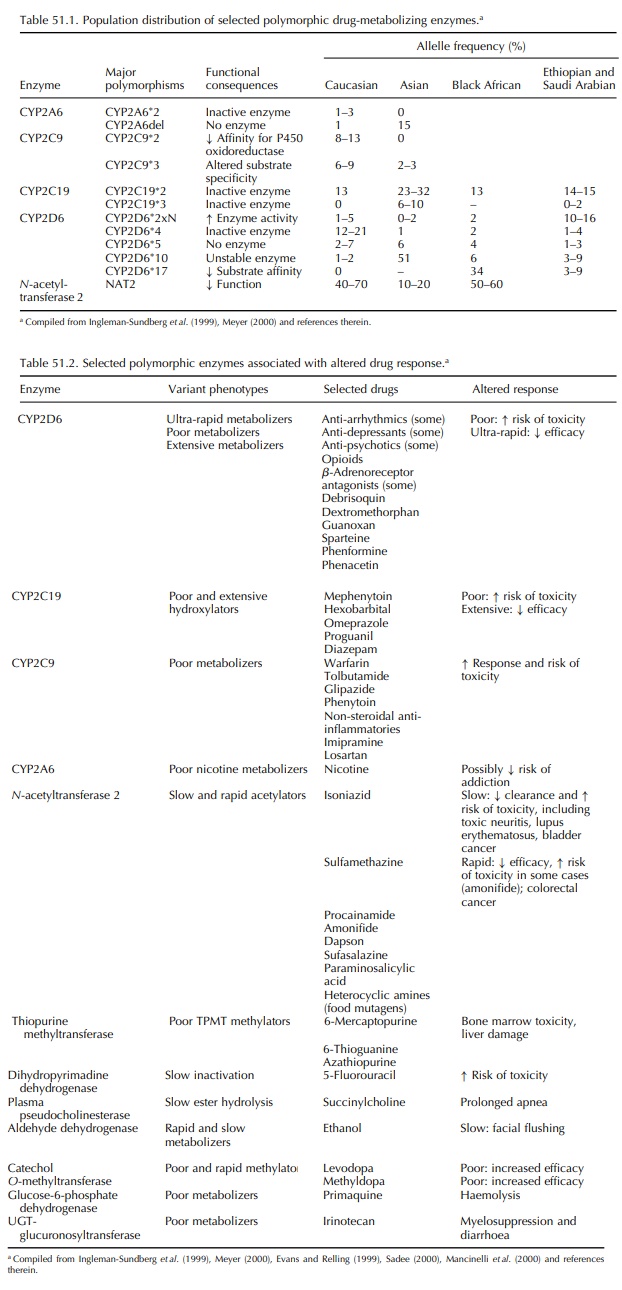
Table 51.2 summarizes some clinically significant polymorphisms affecting drug
metabolism and the drugs and drug effects associated with them; a comprehensive
summary is available at http://www.hapmap.org/cgi-perl/gbrowse/gbrowse.
Continuously updated descriptions of these alle-les and accompanying references
can be found at http://www.imm.ki.se/CYPalleles/.
CYP2D6,
which encodes debrisoquin hydroxylase, was the first of these enzyme-coding
genes to be cloned and characterized, and it remains among the most studied. It
is involved in the metabolism of many commonly used drugs, including tricyclic
anti-depressants, neuroleptics, anti-arrhythmics and other cardiovascular drugs
and opioids. Variant alleles may differ from the wild-type (normal) gene by one
or more point mutations, gene deletions, duplications, multiduplications or
amplification. These may have no effect on enzyme activity or may code for an
enzyme with reduced, absent or increased activ-ity. The genetics and related
biochemistry of these pathways are still being elucidated and are more complex
than the following simplistic descriptions imply. Extensive metabolizers, representing 75%– 85% of the general
population, are homozygous or heterozygous for the wild-type, normal activity
enzyme. Intermediate (10%–15% of the
population) and poor (5%–10%) metabolizers carry two reduced or
loss-of-activity alleles. These individuals are likely to exhibit increased
drug plasma concentrations when given standard doses of drugs are metabolized
by this enzyme; this functional overdose results in increased risk of
dose-dependent ADRs associated with these drugs. These individuals also are
likely to experience lack of efficacy with prodrugs that require activa-tion by
this enzyme; lack of morphine-related anal-gesic response to the prodrug
codeine is one example. Ultrarapid
metabolizers (1%–10%) carry duplicated
or multiduplicated active genes; they will metabolize some drugs very
rapidly, never achieving a therapeu-tic plasma drug concentration (and hence
expected efficacy) at a standard dose. Alternately, an ultra-rapid
metabolizer-given codeine may experience an ADR usually associated with
morphine because of the increased conversion of prodrug to active drug; this
often is true of active metabolites, as well.
Two
variant alleles of CYP2C9, which result in reduced affinity for P450
oxidoreductase or altered substrate specificity, are associated with increased
risk of haemorrhage with standard doses of the anti-coagulant warfarin. The
clearance of S-warfarin in patients who are homozygous for one of the
polymorphisms is reduced by 90% compared with patients who are homozygous for
the wild-type allele (Ingleman-Sundberg et
al. 1999). Similar reduc-tions in drug clearance related to one of these
polymorphisms have been documented with other CYP2C9 substrates such as
ibuprofen and naproxen (non-steroidal anti-inflammatories), phenytoin
(anti-epileptic), tolbutamide (hypoglycemic/anti-diabetic) and losartan
(angiotensin II receptor antagonist) (Daly, 1995). The high frequency of these polymor-phisms
(up to 37% of one British population was heterozygous for one mutant CYP2C9
allele) and the severity of the potential ADR (haemorrhage with warfarin
treatment) make this an important consider-ation in the selection and dose of
warfarin and other affected drugs.
A
second important polymorphism affecting the safety of warfarin was reported by
Rieder et al. (2005). They reported
that variants in the gene encoding Vitamin K epoxide reductase complex 1
(VKORC1) explained 25% of the variance in warfarin dose. The effect was three
times that of CYP2C9.
Patients
who are homozygous for the null allele of CYP2C19 (poor metabolizers) are
extremely sensitive to the effects of omeprazole (anti-ulcer), diazepam
(anti-anxiolytic), propranolol (3-blocker), amitripty-line (tricyclic
anti-depressant) and other drugs (Touw, 1997). CYP2C19 also is involved in the
oxidation of the anti-malarial prodrug proguanil to cycloguanil, although it is
unknown whether the polymorphism relates to its anti-malarial effects. The frequency
of this polymorphism (3%–6% in Caucasians and 8%–23% in Asians) defines it as
clinically signif-icant. Polymorphic alleles have been identified for several
Phase II (conjugation) enzymes, and many of these are as important in drug
metabolism as those associated with the Phase I (oxida-tion) enzymes discussed
above. N -acetyltransferase 2, sulfotransferases, glucuronosyltransferases,
catechol O-methyltransferase, dihydropyrimidine dehydroge-nase (DPyDH) and
thiopurine methyltransferase (TPMT) are among the Phase II enzymes known to
have clinically significant effects on drug metabolism (Mancinelli, Cronin and
Sadee, 2000); some of these are summarized in Table 51.2. Polymorphisms of
genes coding for these enzymes are particularly rele-vant in cancer
chemotherapy (severe toxicity for homozygotes of null alleles of TPMT with
thiogua-nine and azathioprine treatment and of DPyDH with 5-flourouracil
treatment) and the treatment of Parkin-son’s disease with l-dopa (low
methylators have an increased response to the drug).
POLYMORPHISMS AFFECTING DRUG TRANSPORT
Although
cellular uptake of some drugs occurs through passive diffusion, membrane
transporters also play a role in the absorption of medicines through the
intestines, their excretion into bile and urine and their uptake into sites of
action (such as brain, testes and cardiovascular tissue; tumour cells; synaptic
cleft and infectious microorganisms) (Evans and Relling, 1999). Increasing
attention is being focused on the possible role of polymorphisms of genes
encoding drug transporters, some of which are summarized in Table 51.3.
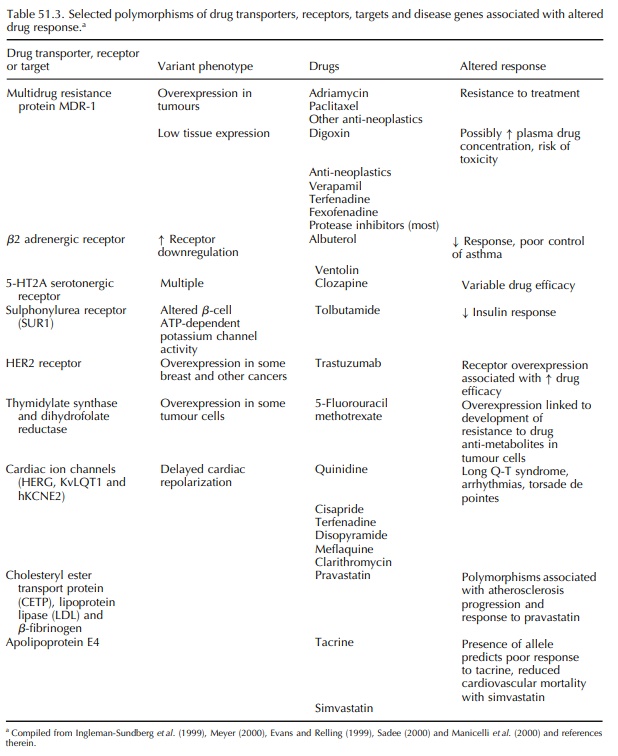
One
example of a transporter with relevance to drug response is p-glycoprotein
(Pgp), an ATP-dependent transmembrane efflux pump that serves to extrude
numerous drugs and other substances out of cells. Pgp is coded for by the
multidrug resistance locus, MDR-1. Hoffmeyer et al. (2000) reported that a specific polymorphism, present in
homozygous form in 24% of their Caucasian sample population, corre-lated with
expression levels and function of MDR-1. Homozygous individuals had
significantly lower MDR-1 expression and exhibited a 4-fold increase in plasma
digoxin concentration after a single oral dose of the drug. Other substrates of
Pgp include impor-tant drugs with narrow therapeutic indices, such as
chemotherapeutic agents, cyclosporin A, verapamil, terfenadine, fexofenadine
and most HIV-1 protease inhibitors (Meyer, 2000). In addition, over-expression
of MDR-1 in cancer tumours has been associated with resistance to adriamycin,
paclitaxel and other anti-neoplastic agents, and additional similar extrusion
pumps are reported to contribute to drug resistance in various tumours (Sadee,
2000). Unfortunately, using Pgp to predict response has not been as successful
as originally hoped.
Another
potentially important gene family with a number of reported variants that may
affect func-tion is that of the biogenic amine transporters, which play a role
in the regulation of neurotrans-mitter concentrations (including serotonin,
dopamine and GABA) in synaptic transmission (Jonsson et al., 1998). These transporters are the direct target recep-tors
for many drugs such as anti-depressants and cocaine; polymorphisms of the
serotonin transporter, in particular, have been associated with the modu-lation
of complex behaviour (Heils, Teufel and Petri 1996) and may play a role in
treatment with specific serotonin transporter inhibitors.
Mutations
in other transporter-like proteins such as the sulfonylurea receptor (SUR) that
regulates ATP-sensitive K+ channels and insulin secretion and nuclear factors
such and hepatocyte nuclear factor-1 alpha and factor-1 beta are being studied
both for their role in aetiology of disease and response to therapy. Pearson et al. (2004) reported on an elegant
study to evaluate the metabolic picture and response to metformin in patients
with type 2 diabetes and maturity onset of the young caused by mutations in
either HNF-1 alpha and HNF-1 beta.
POLYMORPHISMS AFFECTING DRUG RECEPTORS AND TARGETS
Many
drugs interact with specific targets such as receptors, enzymes and other
proteins involved with cell cycle control, signal transduction and other
cellu-lar events. Genes encoding these targets occur in polymorphic forms that
may alter their pharma-cologic response to specific medicines. For exam-ple,
variants affecting β-adrenergic receptors are a major determinant of β-agonist
bronchodilator (e.g. albuterol) response in asthmatic patients. A specific
common polymorphism has been linked to increased β receptor down-regulation in
response to treat-ment with albuterol, which may result in decreased drug
efficacy and duration of action (Tan et
al., 1997; Liggett, 2000). However, other studies have failed to show the
expected correlation between the variant and clinical response (Lipworth et al., 1999).
Drysdale
et al. (2000) suggested that specific
haplo-types (the array of alleles on
a given chromo-some) may have greater predictive value regarding response to
β-agonist bronchodilators than the pres-ence of individual polymorphisms. They
reported marked variation in the ethnic distribution of the most frequently
observed haplotypes (>20-fold differences) and in the mean β-agonist
responses by haplotype pair (>2-fold differences). These authors suggested
that the interactions of multiple polymorphisms within a haplotype may affect
biologic and therapeutic pheno-types and that haplotypes may be useful as
pharma-cologically relevant predictive markers.
Arranz
et al. (2000) completed a
comprehen-sive study of variants in multiple neurotransmitters and receptors in
200 schizophrenic patients. They reported that a set of six sequence variants
involv-ing the 5-hydroxytryptamine (serotonin) receptor, the histamine receptor
(H2) and the promoter region of the serotonin transporter gene successfully
predicted response to treatment with clozapine (a neuroleptic) in 76% of
patients, with a sensitivity of 95% for satis-factory response. Several of
these individual polymor-phisms had been previously studied in this context,
but with inconsistent findings. If the results of this retrospective study are
prospectively validated, then they will form the basis of a simple test to
optimize the usefulness of this expensive drug in a heteroge-neously responsive
group of patients.
The
risk of drug-induced long QT syndrome, a cause of sudden cardiac death in
individuals with-out structural heart disease, has been linked to five gene
variants, each encoding structural subunits of cardiac ion channels that affect
sodium or potas-sium transport and are affected by anti-arrhythmics and other
drugs (Priori et al., 1999). Priori et al. (1999) reported that a
significant number of indi-viduals carry ‘silent mutations’ of these genes; the
resulting alterations are insufficient to prolong the QT interval at rest, but
affected individuals may be especially sensitive to any drug that affects
potassium currents. The combination of these silent mutations with even modest
blockade induced by a variety of drugs used for many purposes can result in
prolonga-tion in action potential that is sufficient to trigger the onset of a
serious ventricular arrhythmia (torsade de pointes). Roden and his colleagues,
however, found less than 10% of patients suffering from drug-induced long QT
actually had any of the known mutations associated with familial long QT
syndrome (Yang et al., 2002).
Polymorphisms
affecting steroid hormone nuclear receptors may affect individual response to
drugs and hormones. For example, glucocorticoid resis-tance in asthma patients
has been associated with increased expression of the glucocorticoid recep-tor
3-isoform (Sousa et al., 2000);
activating muta-tions of the mineralocorticoid receptor have been linked to
hypertension exacerbated by pregnancy (Geller et al., 2000) and dominant negative muta-tions of peroxisome
proliferator-activated receptor gamma (PPAR gamma) have been associated with
severe insulin resistance, diabetes mellitus and hyper-tension. Huizenga et al. (1998) identified a poly-morphism
affecting the glucocorticoid receptor that was present in 6% of their elderly
study popula-tion. These individuals appeared healthy but exhibited increased
sensitivity (reflected in cortisol suppres-sion and insulin response) to
exogenously adminis-tered glucocorticoids. The authors postulated that this
increased lifelong sensitivity to endogenous gluco-corticoids might be
reflected in the observed trends towards increased body mass index and
decreased bone mineral density in affected individuals. This polymorphism also
may be related to the development of early or serious ADRS with exogenous
glucocor-ticoid treatment in carriers, but this has not yet been established.
Some
investigators have reported a relationship between variants in the angiotensin
converting enzyme (ACE) gene and individual sensitivity to ACE inhibitors such
as enalapril, lisinopril and captopril, but the results reported by other teams
fail to show an association, so this finding remains to be confirmed (Navis et al., 1999).
The
beta adrenergic receptor is the target for drugs used to treat asthma,
hypertension, and heart failure. Two polymorphisms appear to have an effect on
some drugs for the treatment of asthma as well ask risk of heart failure.
Small
et al. (2002) reported that African
Americans were at significantly greater risk of developing heart failure if
they carried a single copy of the 2c dele-tion of the adrenergic receptor. In
animal models, this deletion results in an ineffective form of the recep-tor
and higher norepinephrine levels. When combined with the ‘hyperfunctioning’ 389
mutation, the risk was multiplied several fold. The 2c deletion is more common
in African Americans, and the authors hypothesize that this may be the reason
for higher rates of heart failure in African Americans. The numbers in the
study were smaller for Caucasians and did not result in a statistically
significant risk. Hajjar and MacRae (2002) in their editorial accompanying this
paper warn that the data must be replicated to be considered.
POLYMORPHISMS RELEVANT TO CANCER CHEMOTHERAPY
The
basis of many forms of cancer chemotherapy involves the administration of
maximum tolerated dosages with the goal of inflicting the greatest damage to
malignant cells while causing the least damage to normal tissue. Genetic
variations of drug-inactivating enzymes in normal tissues may increase the risk
of severe toxicity or even death. As mentioned above, TMPT-deficient
(homozygous; –0.3% of the popu-lation) individuals treated for acute
lymphoblastic leukaemia with standard doses of mercaptopurine, thioguanine and
azathioprine (immunosuppressant) may experience severe and potentially lethal
bone marrow toxicity. A dose reduction of up to 15-fold may be needed to avoid
haematotoxicity in these patients (Evans et
al., 1991). TPMT genotyping or phenotyping (by assessing red blood cell
enzyme levels) before the institution of therapy with any of these agents has
become accepted practice at some medical centres (Sadee, 2000).
Several
similar examples have been documented (Iyer and Ratain, 1998): patients with
variant DPyDH cannot inactivate 5-fluorouracil, resulting in myelo-suppression
and neurotoxicity, while overexpression of DPyDH in tumours is linked to
resistance to that drug; N-acetyltransferase-2 rapid acetylators (30%– 60% of
Caucasians and 80%–90% of Asians) are at risk of greater bone marrow toxicity
with amon-afide treatment (topo-isomerase II inhibitor), and patients who have
a genetic deficiency of glucuronida-tion because of a variant promoter of
UGT-glucuronosyltransferase UGTIA1 are at increased risk of myelosuppression
and diarrhoea when treated with the topoisomerase I inhibitor irinotecan. At
least one example of an activating
variant of a co-factor/enzyme has been reported: mutations of NAD(P)H (nicotinamide-adenine
dinucleotide phos-phate, reduced form) : quinone oxidoreductase (which
activates cytotoxic anti-tumour quinones such as mito-mycin C) protect against
cytoxic metabolites but also may reduce anti-tumour efficacy (Gaedigk et al., 1998).
Growth
factor receptors may be overexpressed in some tumours, potentially affecting
the efficacy of chemotherapy. One example of this involves the humanized
monoclonal antibody trastuzumab (HerceptinTM , which was designed to
target an onco-gene (HER2/neu) that is overexpressed in some breast cancers and
other cancers with poor prognoses. Trastuzumab, when given with paclitaxel and
doxoru-bicin, enhances the cytotoxic effects of the anti-neoplastic agents in
breast cancer tissues with high HER2/neu expression. Some researchers suggest
that an optimal approach to cancer chemotherapy would involve genotyping both
malignant and normal cells when feasible (Sadee, 2000).
Unfortunately,
the Epidermal Growth Factor Recep-tor (EGFR) story is not as clearcut, but the
use of pharmacogenomics and drug probes are helping scientists to understand
the redundant pathways of growth. Early enthusiasm about the effectiveness of
EGFR inhibitors (erlotinib and gefitinib) was followed by studies that showed
no benefit when combined with cytotoxic drugs. However, Lynch and associates
reported activating mutations in the EGF receptor that appeared to underlay
responsiveness of non-small cell lung cancer to gefitinib (2004). A subgroup of
patients had impressive response: women, patients who had never smoked,
patients with adenocarci-noma, and Asians. A majority of the tumours in these
patients were found to have a mutation in the EGFR gene which increased the
sensitivity of the tumour to anilinoquinazoline inhibitors of EGFR. This is a
rapidly moving and potentially fruitful area of both basic and clinical
research.
Adoption
of predictive tests associated with drug treatment has been extremely high in
oncology. The percentage of physicians using a predictive test prior to
treatment with Herceptin has exceeded early esti-mates and sales of Herceptin
have exceeded expecta-tions. Clearly the use of a predictive test was a benefit
to doctors, patients, and the developers of Herceptin, Genentech.
Related Topics
