Interactions in Relation to Multiple Drug Prescribing
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Drugs and the Elderly
Adverse drug reactions (ADRs) are common in the elderly. This is frequently a consequence of multiple drug prescribing which leads to the occurrence of drug–drug interactions.
INTERACTIONS IN RELATION TO
MULTIPLE DRUG PRESCRIBING
Adverse
drug reactions (ADRs) are common in the elderly. This is frequently a
consequence of multiple drug prescribing which leads to the occurrence of
drug–drug interactions. The risk of potentially inap-propriate drug
combinations is also increased by the greater number of physicians prescribing
medications for an elderly patient (Tamblyn et
al., 1996). Drug interactions represent a change in either the magnitude or
the duration of action of one drug caused by the presence of the second. This
may enhance or reduce the efficacy of one or both of the drugs or a new effect
may appear which is not seen with either of the drugs alone. Interactions may
be pharmacokinetic or pharmacodynamic. The most important adverse inter-actions
occur with drugs that have easily recognisable toxicity and a low therapeutic
index (i.e. the dose or plasma concentration of drug which is effective lies
close to that which causes toxicity) (Lin and Lu, 1998). Current knowledge of
drug–drug interactions has increased dramatically over the recent years, but
studies rarely involve the frail elderly who will be more susceptible to the
adverse effects of interacting medications. There are additional concerns about
interactions of drugs with herbal supplements, certain food stuffs and alcohol,
which are also important to consider when prescribing.
PHARMACOKINETIC INTERACTIONS
Drug
interactions may result in impaired drug absorp-tion from the gastrointestinal
tract. The rate at which a drug is absorbed may be decreased by drugs such as
anticholinergics, which inhibit gastric motility; conversely, drugs such as
metoclopramide (which increase gastric motility) may enhance the absorp-tion
rate. Certain drugs form chelates and complexes with other drugs, altering
their solubility and absorp-tion. For example, agents that bind to digoxin in
the gut (such as antacids and cholestyramine) reduce the extent of its
absorption by 20%–35% (Brown and Juhl, 1976). However, despite these potential
interac-tions few drug–drug interactions affect drug absorp-tion to a
clinically significant extent (May, Dipiro and Sisley, 1987; McInnes and
Brodie, 1988). Drugs that undergo extensive first-pass metabolism may be
affected by other drugs, which alter liver blood flow or compete for
metabolism. For example, the non-selective monoamine oxidase inhibitors
(MAOIs), such as phenelzine, reduce the first-pass metabolism of tyramine
(found in cheese, tomatoes and choco-late), pseudoephedrine (in cough mixtures)
and many other direct and indirect sympathomimetic agents (Tollefson, 1983). As
a result, large amounts of these amines reach the sympathetic nervous system,
where they stimulate the interneuronal release of nora-drenaline. MAOI prevents
noradrenaline breakdown, producing a syndrome of sympathetic over-activity
characterised by headache, hypertension, excitement and delirium (Tollefson,
1983).
Drugs
may also affect the distribution of others within the body. When two or more
highly protein-bound drugs are administered concurrently, compet-itive binding
by one may increase the free fraction or unbound portion of the other. The
importance of this interaction has probably been overstated. For example, the
NSAIDs may displace warfarin from its binding site and increase its
anticoagulant effect, but this effect is negligible in vivo (O’Callaghan, Thomp-son and Russell, 1984); it is much more
likely that the NSAIDs inhibit warfarin metabolism (O’Reilly et al., 1980). Similarly,
tolbutamide-induced hypogly-caemia with the addition of azapropazone has been
reported (Waller and Waller, 1984). Although the interaction may have been due
to displacement of the oral hypoglycaemic agent from albumin leading to
enhanced hypoglycaemia, inhibition of tolbutamide metabolism by the NSAID was
probably more impor-tant (Andreasen et al.,
1981).
Inhibition
or induction of drug metabolism is one of the most important mechanisms for
drug–drug inter-actions. Interactions involving a loss of action of one of the
drugs are at least as frequent as those involv-ing an increased effect (Seymour
and Routledge, 1998). There are many examples of one drug inter-fering with the
metabolism of another by inhibition of the cytochrome P450 (CYP) enzymes in the
liver (Tanaka, 1998). The enzymes responsible for trans-forming drugs in humans
belong to six CYP subfam-ilies, that is CYP1A, 2A, 2C, 2D, 2E and 3A. Each
subfamily contains a number of different isoforms. It has been estimated that
about 90% of human drug oxidation can be attributed to six of these, that is
CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A, and enzyme inhibition
interactions have been reported with all (Kinirons and Crome, 1997; Seymour and
Routledge, 1998). Each CYP isoenzyme may metabolise many drugs, so the
potential for drug– drug interactions is high in patients taking several
medications (Lin and Lu, 1998). For example, in a group of elderly male
patients, cimetidine inhibited the metabolism of procainamide, giving rise to
toxic plasma concentrations of the antiarrhythmic (Bauer, Black and Gensler,
1990).
Other
drugs which are similarly affected by cime-tidine are benzodiazepines,
-adrenoceptor blockers, tricyclic antidepressants, theophylline, phenytoin and
oral anticoagulants. Although few of these drug–drug interactions are of
clinical significance (Sax, 1987), caution is indicated when cimeti-dine is
given concomitantly with drugs that have a narrow range of therapeutic
concentration such as warfarin, theophylline and phenytoin: in one study, 2
days of cimetidine therapy decreased theophylline clearance by 39% (Jackson et al., 1981). Other common inhibitors
of one or more CYP isoenzymes include amiodarone, fluconazole, erythromycin,
clar-ithromycin, sulphonamides, ciprofloxacin, omepra-zole and paroxetine.
Occasionally, clinically severe interactions can occur as has been shown with
combined administration of terfenadine and ketocona-zole (Honig et al., 1993; Monaghan et al., 1993) and erythromycin (Honig et al., 1992) and itraconazole
(Pohjola-Sintonen et al., 1993),
resulting in prolonga-tion of the QT interval and torsade des pointes. At
present, there is no evidence that CYP inhibition by these agents is affected
by age (Kinirons and Crome, 1997). There is increasing evidence that many
herbal remedies and other dietary supplements may have pharmacological activity
that can lead to potential interactions with drugs when taken together. Many of
these interactions have been identified to occur by inhibition of CYP
isoenzymes although the effects of herbal supplements, diet and food on drug
metabolism require further study in older persons (Kinirons and O’Mahony, 2004).
Liver
enzyme induction by one drug may lead to inactivation of the second drug.
Well-recognised examples include the decreased efficacy of warfarin seen with
barbiturate therapy and the reduced efficacy of dihydropyridine calcium-channel
blocking drugs with carbamazepine therapy (Capewell et al., 1988). The delay between the commencement of the
enzyme-inducing agent and its full effect can take 7–10 days, making
recognition of the interaction more difficult (Seymour and Routledge, 1998).
However, in general terms, elderly individuals appear to be less sensitive to
drug induction than younger individuals (Lin and Lu, 1998). For example, the
distribution of hexobarbitol before and after treatment with rifampicin was
studied in young and elderly volunteers. Rifampicin produced differential
increases in hexobarbitol metabolism with 90- and 19-fold increases in the
young and elderly volunteers, respectively (Smith et al., 1991).
Finally,
drug–drug interactions may occur in the kidney resulting in altered drug elimination.
This subject has been recently reviewed by Bonate, Kelly and Weir (1998), who
concluded that clinically signif-icant drug interactions due to a renal
mechanism are relatively rare. Five potential mechanisms exist for drug
interactions in the kidney (Table 42.3), and the best recognised is competitive
inhibition of tubular secretion leading to an increase in drug concentration.
An example of this interaction is the co-administration of probenecid with
penicillin. Non-competitive interference with drug secretion may also occur,
for example prolonged treatment with thiazide diuretics causes a compensatory
increase in proximal tubule reabsorption of sodium, resulting in increased
lithium reabsorption (Peterson et al.,
1974). This interaction has resulted in serious lithium toxi-city due to
lithium accumulation (Mehta and Robin-son, 1980). NSAIDs also decrease the
renal elimi-nation of lithium by up to 60%, but the mechanism is uncertain
(Amdisen, 1982; Jefferson et al.,
1986). Similarly, the administration of quinidine results in an increase in the
plasma concentration of digoxin in over 90% of patients (Bigger, 1982).
Although this is partly due to displacement of digoxin from its binding sites
in tissues, its renal clearance is reduced by 40%–50% with regular
administration of quinidine. Similar interactions have been reported with
amiodarone (Moysey et al., 1981;
Oetgen et al., 1984) and verapamil
(Pederson et al., 1983), leading to
70%–100% increases in serum digoxin concen-trations. Although the precise
mechanisms have not been elucidated, recent reports suggest that inhibi-tion of
ATP-dependent P-glycoprotein-mediated drug transport in renal tubular cells
(Inui, Masuda and Saito, 2000) by verapamil and quinidine may lead to decreased
renal tubular elimination of digoxin (Fromm et
al., 1999; Verschraagen et al.,
1999).

Drug
transport systems may be important in tissues other than the kidney and are
found in the membrane of epithelial cells in the intestinal wall and
blood-brain barrier. P-glycoprotein plays an active role in the uptake and
efflux of many substrates including vari-ous drugs. Polypharmacy may have
specific relevance for elderly patients treated with substances that affect
drug transporters due to adverse interactions (Kinirons and O’Mahony, 2004).
For example, ciclosporin is a substrate of both CYP3A and P-glycoprotein; other
drugs affecting either mechanism may alter its phar-macokinetics (Kovarik and
Koelle, 1999). Despite this, the available pharmacokinetic data suggests that
no dose modification of ciclosporin is required in the elderly.
PHARMACODYNAMIC INTERACTIONS
An
antagonistic pharmacological interaction between two drugs may counteract the
intended therapeutic effects. For example, co-administration of NSAIDs and
antihypertensives may lead to a reduced hypoten-sive effect due to sodium
retention by the anal-gesics. One Australian study found that 12% of almost
3000 non-institutionalised elderly patients studied were taking NSAIDs and
anti-hypertensive medica-tion simultaneously. Furthermore, NSAID usage was an
independent risk factor for hypertension in this age group (Johnson et al., 1993).
Finally,
indirect pharmacodynamic effects may occur when one drug’s pharmacological
effect influences another drug’s action. For exam-ple, enhanced myocardial
depression, hypotension and atrioventricular block may occur when -
adrenoceptor blockers are administered with vera-pamil or diltiazem (Krikler
and Spurrell, 1974; Edoute et al.,
2000).
Despite the many ways in which drug–drug inter-actions may
occur, it is likely that only about 10% of potential interactions result in
clinically signifi-cant events. However, while death or serious clinical
consequences are rare, low grade, clinical morbidity in the elderly may be much
more common (Seymour and Routledge, 1998). Non-specific complaints such as
confusion, lethargy, weakness, dizziness, incon-tinence, depression and falling
may indicate an underlying drug–drug interaction. The drug interac-tions of
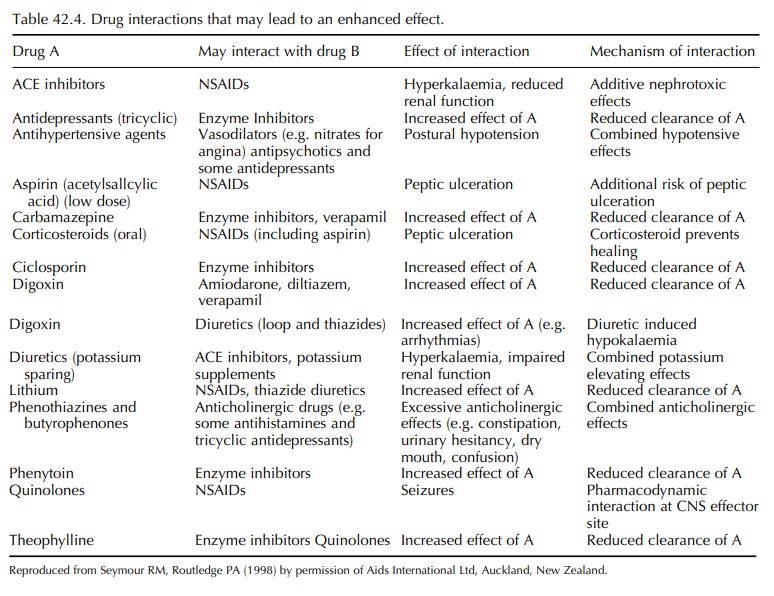
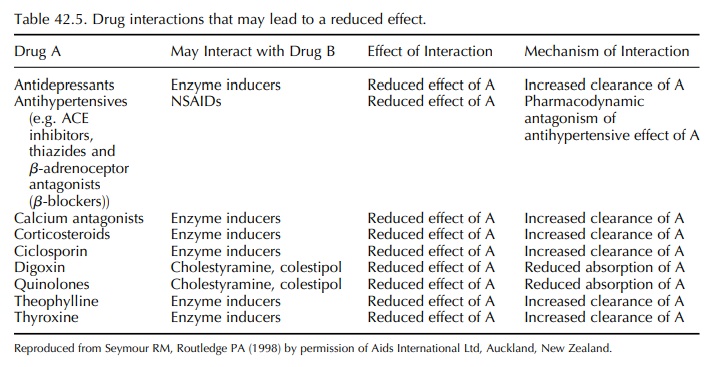
clinical importance in the elderly have been reviewed by Seymour and Routledge
(1998) and are listed in Tables 42.4 and 42.5. In some cases, the cause of the
interaction is complex, involving both pharmacokinetic and pharmacodynamic mechanisms.
For example, epileptic patients with psychiatric co-morbidity may be
particularly vulnerable because of combined use of psychotropic and
antiepileptic drugs. In particular, antidepressants and antipsychotic drugs are
believed to lower the seizure threshold (Alldredge, 1999; Coleman, 2004). In
general, the potential for drug–drug interactions in psychiatric patients is
high because of the need for combined therapy to treat co-morbid psychiatric
disorders, to treat the adverse effect of a medication or to treat concomitant
medical conditions. In particular, the selective serotonin uptake inhibitors
fluoxetine and paroxetine are potent inhibitors of CYP2D6 and have the
potential to increase the plasma concentra-tions of many of the major tranquilisers,
including haloperidol and thioridazine; fluvoxamine inhibits the metabolism of
many of the benzodiazepines (Sproule et
al., 1997).
Related Topics
