Risk Management Guidances
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: US Activities in Risk Management of Pharmaceutical Products
The Prescription Drug User Fee Act of 2003 (PDUFA III )specifically addressed risk management, noting that efficient risk management as one of FDA’s five strategic goals, including both the new drug review process and oversight after approval.
RISK MANAGEMENT GUIDANCES
The
Prescription Drug User Fee Act of 2003 (PDUFA III )specifically addressed risk
management, noting that efficient risk management as one of FDA’s five strategic
goals, including both the new drug review process and oversight after approval.
Acknowledg-ing that it is impossible at the time of approval to know everything
about a medicine’s safety, PDUFA III mandated that there be increased
surveillance of the safety of medicines during their first 2 years on the
market (or first 3 years for drugs with potentially seri-ous safety concerns
identified at the time of approval). The FDA also agreed to develop regulatory
strategies and guidance documents on risk management. Three guidance documents
were developed with input from the public and industry. These guidances,
summa-rized below, were published as final documents in March 2005.
Due
to its relevance to this chapter, the ‘Guidance on Good Pharmacovigilance Practices
and Pharma-coepidemiologic Assessment’ is provided in full at the end of this
chapter.
GUIDANCE ON PRE-MARKETING RISK ASSESSMENT
This regulatory guidance focuses on approaches a industry might consider throughout all stages of the clinical development of products. Some key compo-nents of the guidance include specific recommendations to industry for improv-ing the assessment and reporting of safety during drug development trials;
·
improving the assessment of important safety issues during
registration trials and to provide best practices for analyzing and reporting
data that are developed as a result of a careful pre-approval safety evaluation
and
·
building on (but not superceding) a number of existing FDA
and ICH (International Conference on Harmonisation of Technical Requirements
for Registration of Pharmaceuticals for Human Use) guidances related to
pre-approval safety assessments.
GUIDANCE ON DEVELOPMENT AND USE OF RISK MINIMIZATION ACTION PLANS
This
guidance provides a conceptual framework on the development, implementation and
evaluation of risk minimization action plans for prescription drug and
biological products. It focuses on (1) initiating and designing plans called
risk minimization action plans or RiskMAPs to minimize identified product risks,
(2) selecting and developing tools to minimize those risks, (3) evaluating
RiskMAPs and monitoring tools, (4) communicating with FDA about RiskMAPs, and
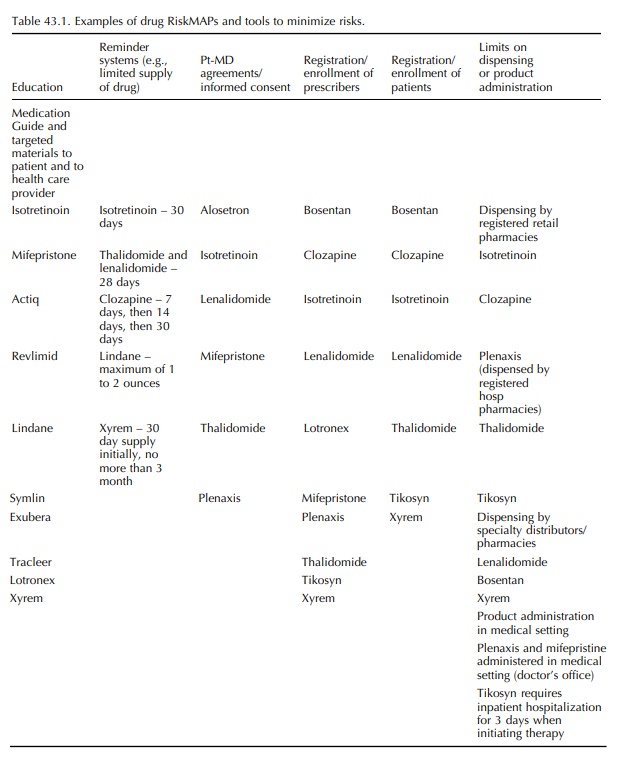
(5) the recommended components of a RiskMAP submission to FDA. Table 43.1
provides recent exam-ples of drug RiskMAPs and tools to minimize risks.
GUIDANCE ON GOOD PHARMACOVIGILANCE PRACTICES AND PHARMACOEPIDEMIOLOGIC ASSESSMENT
This
guidance document focuses on pharmacovig-ilance activities in the post-approval
period. Phar-macovigilance is defined to mean all scientific and data gathering
activities relating to the detection, assessment and understanding of adverse
events. This includes the use of pharmacoepidemiologic studies. These
activities are undertaken with the goal of identi-fying adverse events and
understanding, to the extent possible, their nature, frequency and potential
risk factors.
CONCLUSIONS
Pharmaceutical
risk management faces important challenges in addressing innovative therapies,
public expectations of product safety and optimizing patient selection to
better minimize adverse outcomes. Regu-latory pharmacovigilence activities have
a criti-cal role in assuring product safety by means of proactively designing
and implementing interven-tions to minimize a product’s risks.
Pharmacovig-ilence also provides a framework for evaluating these interventions
in light of new knowledge that is acquired over time and revising interventions
when appropriate.
Related Topics
