Pharmaceutical Water Systems
| Home | | Pharmaceutical Technology |Chapter: Pharmaceutical Engineering: Bioprocessing
In general, pharmaceutical water systems employ combinations of technologies described earlier.
PHARMACEUTICAL WATER SYSTEMS
In
general, pharmaceutical water systems employ combinations of technologies
described earlier. However, since the objective is unique, we review some of
these methods while introducing issues that are specifically related to the
pro-duction of different qualities of water (Kuhlman and Coleman, 1995). Table
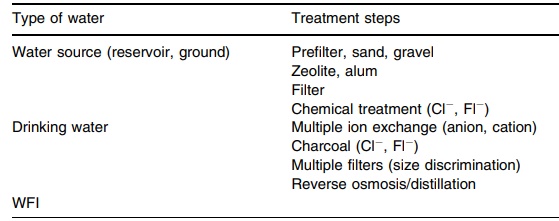
16.1 summarizes water treatments and uses.
Pretreatment and Sources of
Water
Potable
(drinking) water is not suitable for pharmaceutical purposes. The United States
Environmental Protection Agency limits allow 500 recoverable micro-organisms
per milliliter, none of which can be coliform organisms in drinking water.
Drinking water requires further treatment to meet the requirements for use in
pharmaceutical processes.
Water
is pretreated to remove materials likely to be detrimental to the purification
equipment. This pretreatment takes many forms. A multimedia bed (different
gravels in a carbon steel vessel) is used to remove solids from the municipal
water. Common problems include high bacterial or particulate counts in the
effluent. This technique is highly inefficient because the container is susceptible
to corrosion, the media is porous, and the piping contains dead legs, cracks,
and crevices.
Water for Injection
Water
for injection (WFI) is prepared following pretreatment and further
puri-fication, including ion exchange, distillation, and reverse osmosis
(Kuhlman and Coleman, 1995). WFI must contain 50 recoverable bacterial colonies
or less per milliliter for immediate use. Its preparation by distillation or
reverse osmosis renders it sterile, from which it must be protected from
contamination by endotoxins or microorganisms.
TABLE 16.1 Water Treatment
Ion Exchange
Zeolite
water softener is an exchanger that replaces calcium ions with magne-sium ions.
Regeneration of the resin is necessary and usually conducted with brine. Consequently,
chloride ions that attack certain types of composite membranes may enter the
feedwater stream. Bacteria may also propagate in this system.
Activated
carbon filters employ a carbon steel tank filled with gravel and covered with
activated charcoal (anthracite). Again, this is a source of bacteria and
chloride ions. Deionized water is produced by passing treated water through a
mixed-bed or a two-bed cation/anion exchange resin system. The resulting water
is deionized because hydrogen ions replace cations and hydroxyl ions replace
anions. Deionized water has little or no bacteria and is easily regenerated.
The potential for microbial contamination during some of these purification
procedures renders additional steps necessary to prepare water suitable for
pharmaceutical processing.
Distillation
Distillation
separates water from other soluble and insoluble components by elevating the
temperature to that at which vapor forms (100○C) in a boiling
chamber and then condensing the vapor into a receiving vessel. The nature of
hydrogen bonding of water imparts a unique property to water. Although it can
be raised to 100○C
with a relatively small amount of energy (80 kcal), it takes almost seven times
this amount (540 kcal) to break the hydrogen bonds and release the water as
steam at the same temperature. Consequently, in the con-densation phase, eight
times as much water at 5○C
(refrigeration temperature) is required to condense the water as steam. These
large exchanges of heat may be used in an efficiently designed still to heat up
water entering a second still. Alternatively, the combined gas law can be
utilized by compressing vapor and therefore elevating its temperature (vapor
compression still).
Reverse Osmosis
Reverse
osmosis units vary in design, construction materials, and membrane type more
than any other unit in the pretreatment process. Usually it is a sin-glepass
system (may not eliminate chlorides). Transmembrane pressures must be
maintained. Osmosis is the process whereby a solution separated from pure water
by a semipermeable membrane induces movement of water toward the region of high
solute concentration. This would ordinarily give rise to an osmotic pressure.
If pressure is applied against the osmotic pressure head, the flow of water can
be induced in the opposite direction, thereby reversing osmosis. This process,
which may be regarded as a form of filtration, removes materials of sizes down
to 200-Da molecular weight in a sequence that usually removes particulates and
viable microorganisms and contaminates molecules sequentially according to size
(i.e., large particles, bacteria, viruses, pyrogens, and ions). Softened
pH-adjusted water is used to maximize the efficiency of ion removal. The ionic
radius affects ion removal, with multivalent ions more readily removed than
monovalent ions.
Storage and Distribution
The
water temperature at the point of use must be such that the water can be
handled without risk. A recirculating ambient loop or a heat exchanger at the
point of use may be required. A sophisticated system of loops and heat
exchangers is required to elevate the water temperature before it returns to a
storage tank. One approach is to maintain an ambient loop during the day and
heat the water during the night. If the water is maintained at ambient
temper-atures for not more than 24 hour, the conditions do not violate current
good manufacturing practice (cGMP) regulations.
Quality Control
Conductivity
and resistivity are convenient online measures that ensure water quality. As it
circulates, water loses resistivity, stabilizing at about 5 MΩ/cm. Some corrosion may take place
in the distribution system, which may ultimately lead to adulteration of the
water. Endotoxin levels are monitored by sampling. Sampled water may be
subjected to the limulus amebocyte lysate test to measure the presence of
endotoxin. This in vitro assay was predated by rabbit pyrogen testing, which
involves monitoring the rabbit’s core body temperature in response to injection
with a water sample. Endotoxin may cause mild immune responses that will be
detected by an increase in body temperature.
Validation
Validation
of any process is required in pharmaceutical manufacturing. The validation
master plan outlines the required content and method of preparing validation
documentation. Validation is integral to the start-up of the entire plant.
Three major sections of the validation procedure are the following:
1. Installation qualification (IQ): establishes and
documents that the unit or system was installed correctly per the
manufacturer’s specification
2. Operational qualification (OQ): establishes and documents
that the unit or system operates as intended
3. Production qualification (PQ): establishes and documents
that the unit or system can fulfill its intended purpose on a reproducible
basis when challenged with realistic worst-case conditions
The
master plan should include a listing of documentation included in validation
files for each system (reference files, vendor data, calibration reports,
standard operating procedures, and inventories). Critical path schedules,
man-power estimates, operator responsibilities, auditing procedures, and
outside validation resources should be included in validation documentation.
Outside validation resources should be recruited. They may include purchase of
vali-dation protocols from commercial vendors, acquisition of data on
validation exercises from equipment vendors, use of testing laboratories for
performance qualification, contracting with other qualified agencies to perform
water sam-pling, and, in the extreme case, contract with a qualified agency to
perform the entire validation exercise (including writing protocols and
performing valida-tion testing). The scale of operation and internal resources
dictate which option to select.
