Antibiotics Assays - Examples of Pharmaceutical Microbial Assays
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Microbiological (Microbial) Assays: Antibiotics-Vitamins- Amino Acids
The microbial assays of ‘antibiotics’ are usually carried out by two standard methods as per Indian Pharmacopoea.
EXAMPLES
OF PHARMACEUTICAL MICROBIAL ASSAYS
The microbial assays have been effectively
extended to a plethora of pharmaceutical
preparations i.e., the secondary
pharmaceutical products. However, this particular section will deal with only the following three types of such products, namely :
(1) Antibiotics,
(2) Vitamins,
and
(3) Amino
Acids.
Antibiotics Assays
The
microbial assays of ‘antibiotics’
are usually carried out by two
standard methods as per Indian Pharmacopoea (1996), namely :
Method A i.e., the ‘Cylinder-Plate Method’ as discussed in Section 10.1.3.1 and
Section 10.3.1.
Method B i.e., the ‘Turbidimetric Method’ as described in Section 10.1.3.2.
A
comprehensive account of the ‘Antibiotic
Assays’ shall now be dealt with under the following sub-heads :
Standard Preparation and Units of Activity
Standard preparation may be
defined as— ‘the authentic sample of the
appropriate antibi-otic for which the potency has been precisely determined
with reference to the appropriate inter-national standard’
However,
the potency of the standard preparation
may be duly expressed either in Interna-tional
Units (IU) or in μg.mg–1 with
respect to the ‘pure antibiotic’.
Important Points
(1) Standard Preparation for India
are adequately maintained at the Central Drugs Labora-tory, Kolkata. A
unit referred to in the ‘official assays’ and ‘tests’ refers to the
specific activity con-tained in such an amount of the respective standard preparation as is duly
indicated by the Ministry of Health and Family Welfare, Government of India
from time to time.
(2) Standard Preparation may be
suitably replaced by a ‘working
standard’ prepared by any laboratory
that must be compared at definite intervals under varying conditions with the ‘standard’.
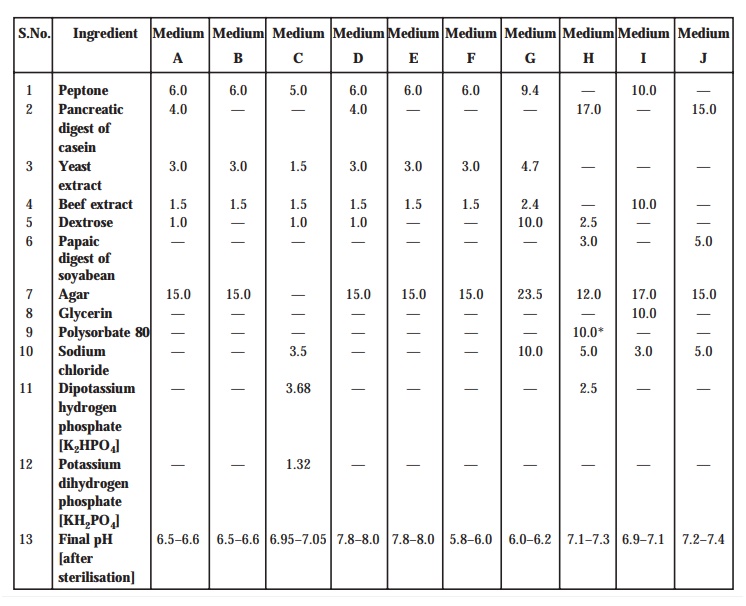
[A] Media. The media necessarily required for the preparation of ‘test organism inocula’ are duly made from the various ingredients
as listed specifically in Table : 10.1. However, one may make minor
modifications of the individual ingredients as and when required or ‘reconstituted dehydrated media’ may be employed provided the resulting media have either almost
equal or definitely better growth-promoting
characteristic features, and ultimately give a similar standard curve-response.
Method : Dissolve the various prescribed
ingredients in sufficient distilled water (DW) to pro-duce 1L, and add
sufficient 1M sodium hydroxide or 1M hydrochloric acid, as required so
that after sterilization the pH must be as stated in Table : 10.1.
TABLE 10.1. Composition of Media : Quantities in
g.L–1
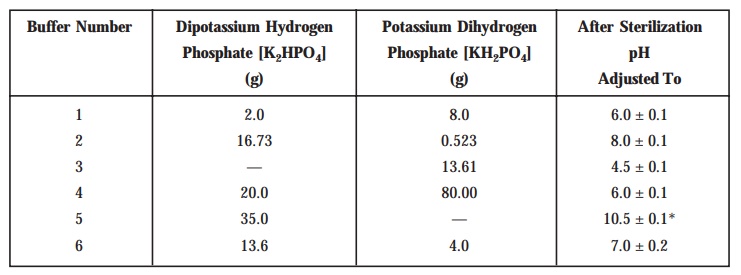
[B] Buffer Solutions : Prepare
the buffer solutions by dissolving
the quantities (see Table 10.2) of K2HPO4
and KH2PO4 in sufficient distilled water to produce 1L
after adjusting the pH with 8 M . H3PO4 or 10 M.KOH. The buffer solutions are
duly sterilized after prepares and the final pH specified in each case must be the one that is
obtained after sterilization.
Table 10.2 : Buffer Solutions
Preparation of Standard Solution
In order
to prepare a ‘Stock Solution’,
dissolve a quantity of the Standard
Preparation of a given antibiotic,
weighed accurately and precisely, and dried previously as duly indicated in
Table 10.3 in the solvent specified in the said Table ; and subsequently dilute
to the required concentration as indicated specifically. It is advisable to
store the ‘Stock Solution’ duly in a
refrigerator (+ 1–5°C), and use within the stipulated period indicated.
On the
particular day intended for carrying out the assay, prepare from the ‘Stock Solution’ at least five or even more test dilutions
whereby the successive solutions increase stepwise in concentra-tion,
invariably in the ratio 1 : 1.25 for
method A or smaller for method B. Use the final diluent
specified and a sequence in a such a manner that the middle or median should
have the concentration as specified duly in Table 10.3.
Preparation of Sample Solution
Based on
the available information for the ‘drug
substance’ under investigation (i.e.,
the ‘un-known’) assign to it an
assumed potency per unit weight or volume, and on this assumption prepare on the day of the assay a ‘Stock Solution’ and test dilution(s) as duly specified for
each individual antibi-otic in Table 10.3, taking particular care to use the same final diluent as employed for the Standard Preparation. The assay with 5 levels of the Standard necessarily requires only one level of the ‘un-known’ at a concentration assumed
very much equal to the ‘median level’ of
the ‘Standard’.
TABLE 10.3 : Stock Solutions and Test Dilutions of
Standard Preparations
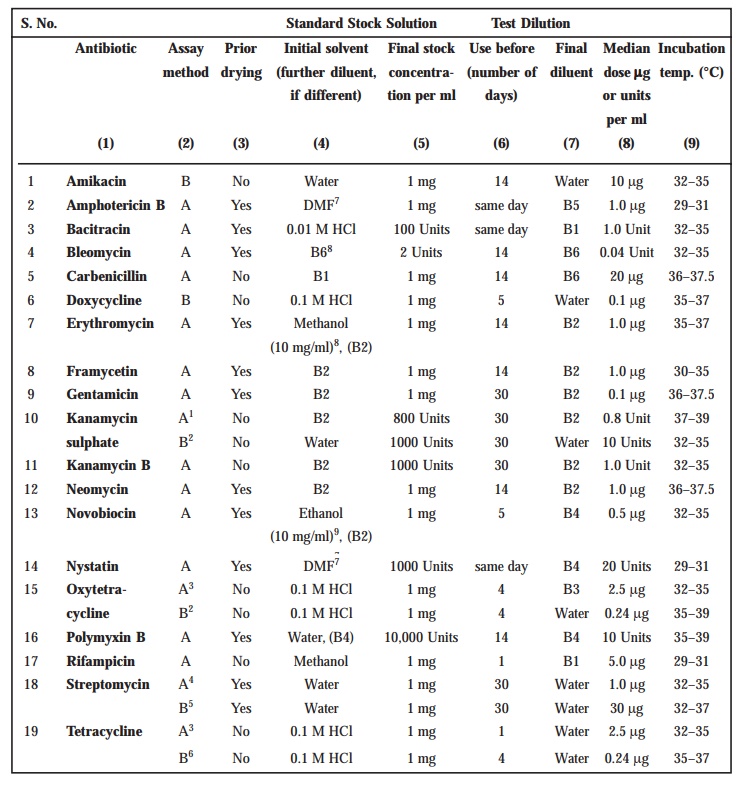
For Amphotericin B, further dilute the stock
solution with dimethylformamide to give concentrations of 12.8, 16, 20, 25 and
31.2 μg per ml
prior to making the test solutions. The test dilution of the sample prepared
from the solution of the substance being examined should contain the same
amount of dimethylformamide as the test dilutions of the Standard Preparation.
For Bacitracin, each of the standard test dilutions
should contain the same amount of hydrochloric acid as the test dilution of the
sample.
For Nystatin, further dilute the stock solution
with dimenthylformamide to give concentrations of 64.0, 80.0, 100.0, 125.0 and
156.0 μg per ml
prior to making the test dilutions. Prepare the standard response line
solutions simultaneously with dilutions of the sample being examined. The test
dilution of the sample pre-pared from the solution of the substance being
examined should contain the same amount of dimethylformamide as the test
dilutions of the Standard Preparation. Protect the soultions from light.
When making the stock solution of Polymyxin B, add
2 ml of water for each 5 mg of the
weighed Standard Preparation material.
Where indicated, dry about 100 mg of the Standard
Preparation before use in an oven at a pressure not exceeding 0.7 kPa at 60°
for 3 hours, except in the fine of Bleomycin (dry at 25° for 4 hours),
Novobiocin (dry at 100° for 4 hours), Gentamicin (dry at 110° for 3 hours) and
Nystatin (dry at 40° for 2 hours).
Where two-level factorial assays are performed use
the following test doses per ml; Amphotericin B, 1.0 to 4.0 μg; Bacteracin, 1.0 to 4.0 Units; Kanamycin
Sulphate, 5.0 to 20.0 units; Streptomycin, 5.0 to 20.0 μg.
[Adapted
From : Indian Pharmacopoea. Vol. II,
1996] 10.6.1.4.
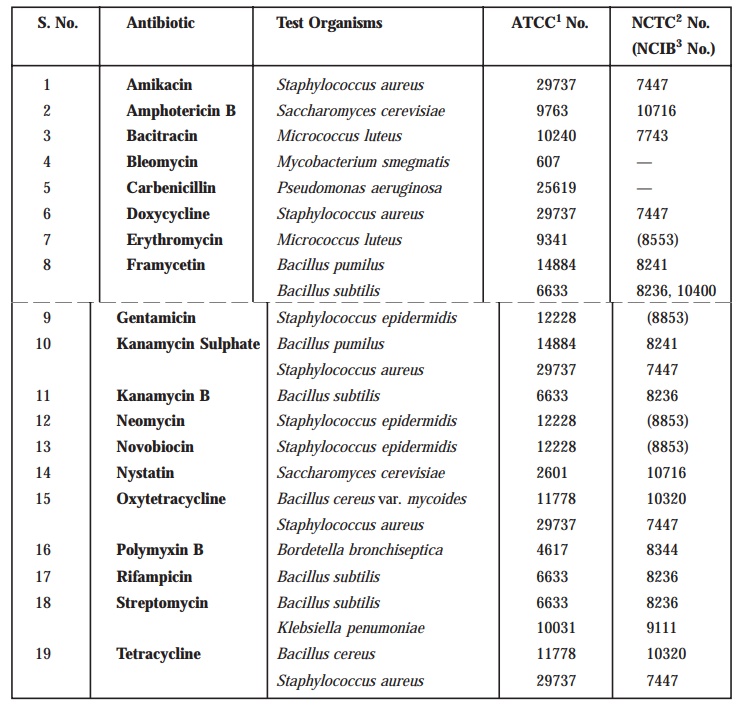
Test Organisms
The
various test organisms for each antibiotic is duly listed in Table
10.4, along with its prop-erly documented identification
number in the following recognized and approved compendia as :
·
American Type Culture Collection (ATCC)
·
National Collection of Type Cultures (NCTC)
·
National Collection of Industrial Bacteria (NCIB).
Usually
maintain a ‘culture’ on the slants
of the medium, and under the specified incubation
conditions as mentioned duly in
Table 10.5, and transfer weekly to fresh slants.
TABLE 10.4 : Test Organisms for Microbiological
Assays of Antibiotics
·
American Type Culture Collection, 21301 Park Lawn
Drive, Rockville, MD 20852, USA.
·
National Collection of Type Cultures, Central
Public Health Laboratory, Colindale Avenue, London NW9 5HT, England.
·
National Collection of Industrial Bacteria, Torry
Research Station, P.O. Box 31, 135 Abbey Road, Aberdeen 98 DC, Scotland.
[Adapted
From : Indian Pharmacopoea, Vol. II,
1996]
TABLE 10.5 : Preparation of Inoculum
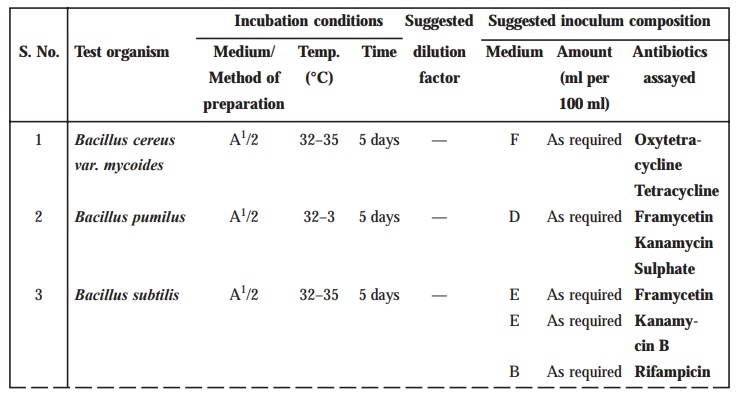
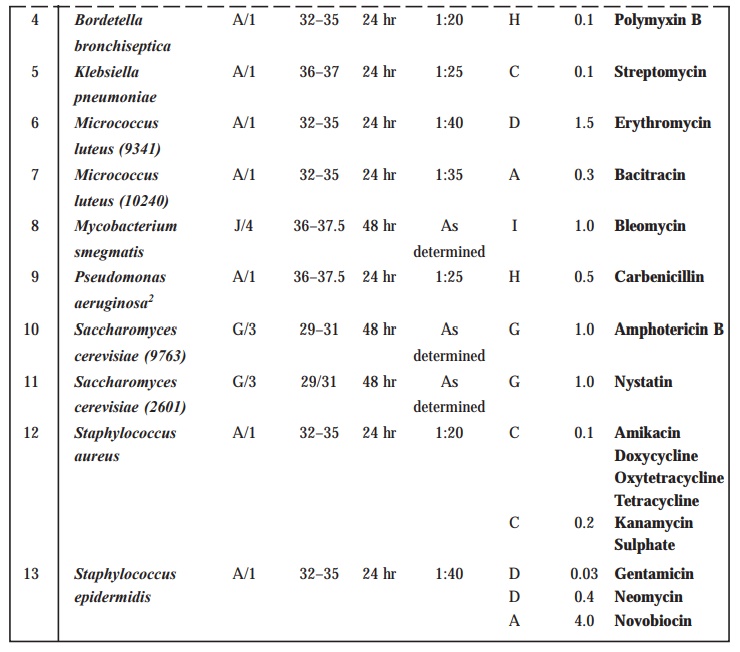
Methods of preparation of test organism suspension
1. Maintain
the test organism on slants of Medium A and transfer to a fresh slant once a
week. Incubate the slants at the temperature indicated above for 24 hours.
Using 3 ml of saline solution, wash
the organism from the agar slant onto a large agar surface of Medium A such as
a Roux bottle containing 250 ml of agar. Incubate for 24 hours at the
appropriate temperature. Wash the growth from the nutrient surface using 50 ml
of saline solution. Store the test
organism under refrigeration. Determine the dilution factor which will give 25%
light transmission at about 530 nm. Determine the amount of suspensions to be
added to each 100 ml of agar of nutrient broth by use of test plates or test
broth. Store the suspension under refrigeration.
2. Proceed
as described in Method 1 but incubate the Roux bottle for 5 days. Centrifuge
and decant the supernatant liquid. Resuspend the sediment with 50 to 70 ml of saline solution and heat the suspension
for 30 minutes at 70°. Wash the spore suspension three times with 50 to 70 ml
of saline solution. Resuspend in 50
to 70 ml of saline solution and
heat-shock again for 30 minutes. Use test plates to determine the amount of the
suspension required for 100 ml of agar. Store the suspension under
refrigeration.
3. Maintain
the test organism on 10 ml agar slants of Medium G. Incubate at 32° to 35° for
24 hours. Inoculate 100 ml of nutrient broth. Incubate for 16 to 18 hours at
37° and proceed as described in Method 1.
4. Proceed
as described in Method 1 but wash the growth from the nutrient surface using 50
ml of Medium 1 (prepared without agar) in place of saline solution.
[Adapted
From : Indian Pharmacopoea, Vol. II,
1996]
Preparation of Inoculum
The
method of preparation of the microbial
suspensions for preparing the inoculum for the assay of various antibiotics is clearly stated in Table 10.5. In an
event when the suspensions are duly prepared
by these methods, one may accomplish and observe that the growth characteristic
features are fairly uniform in order that the inoculum could be determined by
carrying out the following trials.
For Method A. After the suspension is prepared,
as given under Table 10.5, add different
volumes of it to each of several different flasks containing 100 ml of the
medium specified in Table 10.4 (the volume of suspension suggested in Table
10.4 may be used as a guide). Using these inocula, prepare inoculated plates as
described for the specific antibiotic assay. While conducting cylin-der-plate
assays, double layer plates may be prepared by pouring a seed layer (inoculated
with the desired micro-organism) over a solidified uninoculated base layer. For
each Petri dish, 21 ml of the base layer and 4 ml of the seed layer may be
generally suitable. Fill each cylinder with the median concentra-tion of the
antibiotic (Table 10.4) and then incubate the plates. After incubation, examine
and measure the zones of inhibition. The volume of suspension that produces the
optimum zones of inhibition with respect to both clarity and diameter
determines the inoculum to be used for the assay.
For Method B. Proceed as descirbed for Method A
and, using the several inocula, carry
out the procedure as described for the specific antibiotic assay running only
the high and low con-centrations of the standard response curve. After
incubation, read the absorbances of the appropriate tubes. Determine which
inoculum produces the best response between the low and high antibiotic
con-centrations and use this inoculum for the assay.
Apparatus. All equipment is to be thoroughly
cleaned before and after each use. Glassware for holding and transferring test organisms is sterilised by dry heat
or by steam.
Temperature Control
Thermostatic
control is required in several stages of a microbial assay, when culturing a
micro-organisms and preparing its inoculum and during incubation in a plate
assay. Closer control of the temperature is imperative during incubation in a
tube assay which may be achieved by either circulated air or water, the greater
heat capacity of water lending it some advantage over circulating air.
Spectrophotometer
Measuring
transmittance within a fairly narrow frequency band requiers a suitable
spectrophotometer in which the wavelength of the light source can be varied or
restricted by the use of a 580 nm filter
for preparing inocula of the required density, or with a 530 nm filter for reading the absorbance in a tube assay. For the
latter purpose, the instrument may be arranged to accept the tube in which
incubation takes place, to accept a modified cell fitted with a drain that
facilitates rapid change of contents, or preferably fixed with a flow-through
cell for a continuous flow-through analysis. Set the instrument at zero
absorbance with clear, uninoculated broth prepared as specified for the
particular antibiotic, including the same amount of test solution and
formaldehyde as found in each sample.
Cylinder-Plate Assay Receptacles
Use
rectangular glass trays or glass or plastic Petri dishes (approximately 20 ×
100 mm) having covers of suitable material and assay cylinders made of glass,
porcelain, aluminium or stainless steel with outside diameter 8 mm ± 0.1 mm,
inside diameter 6 mm ± 0.1 mm and length 10 mm ± 0.1 mm. Instead of cylinders,
holes 5 to 8 mm in diameter may be bored in the medium with a sterile borer, or
paper discs of suitable quality paper may be used. Carefully clean the
cylinders to remove all residues. An occasional acid-bath, e.g., with about 2M nitric
acid or with chromic acid solution
is needed.
Turbidimetric Assay Receptacles
For assay
tubes, use glass or plastic test-tubes, e.g.,
16 mm × 125 mm or 18 mm × 150 mm that are relatively uniform in length,
diameter, and thickness and substantially free form surface blemishes and
scratches. Cleanse thoroughly to remove all antibiotic residues and traces of
cleaning solution and sterilise tubes that have been used previously before
subsequent use.
Assay Designs
Microbial
assays gain markedly in precision by the segregation of relatively large
sources of potential error and bias through suitable experimental designs. In a
cylinder-plate assay, the essential comparisons are restricted to relationships
between zone diameter measurements within plates, exclu-sive of the variation
between plates in their preparation and subsequent handling. To conduct a
turbidimetric assay so that the difference in observed turbidity will reflect
the differences in the antibi-otic concentration requires both greater
uniformity in the environment created for the tubes through closer thermostatic
control of the incubator and the avoidance of systematic bias by a random
placement of replicate tubes in separate tube racks, each rack containing one
complete set of treatments. The essential comparisons are then restricted to
relationships between the observed turbidities within racks.
Within
these restrictions, two alternative designs are recommended; i.e., a 3-level (or 2-level) factorial
assay, or a 1-level assay with a standard curve. For a factorial assay, prepare
solutions of 3 or 2 corresponding test dilutions for both the standard and the
unknowns on the day of the assay, as described under Preparation of the
Standard and Preparation of the Sample. For a 1-level assay with a standard
curve, prepare instead solutions of five test dilutions of the standard and a
solution of a single median test level of the unknown as described in the same
sections. Consider an assay as preliminary if its computed potency with either
design is less than 60% or more than 150% of that assumed in preparing the
stock solution of the unknown. In such a case, adjust its assumed potency
accordingly and repeat the assay.
Microbial
determinations of potency are subject to inter-assay
variables as well as intra-assay
variables, so that two or more
independent assays are required for a reliable estimate of the potency of a given assay preparation or unknown.
Starting with separately prepared stock solutions and test dilutions of both
the standard and the unknown, repeat the assay of a given unknown on a
different day. If the estimated potency of the second assay differs
significantly, as indicated by the calculated standard error, from that of the
first, conduct one or more additional assays. The combined result of a series
of smaller, independent assays spread over a number of days is a more reliable
estimate of potency than that from a single large assay with the same total
number of plates or tubes.
Methods.
The microbiological assay of antibiotics may
be carried out by Method A or Method B.
[A] Cylinder-Plate or Cup-Plate Method
Inoculate
a previously liquefied medium appropriate to the assay (Tables 10.1 and 10.3)
with the requisite quantity of suspension of the micro-organisms, add the
suspension to the medium at a tempera-ture between 40° and 50° and immediately
pour the inoculated medium into Petri dishes or large rectan-gular plates to
give a depth of 3 to 4 mm (1 to 2 mm for nystatin).
Ensure that the layers of medium are uniform in thickness, by placing the
dishes or plates on a level surface.
The
prepared dishes or plates must be stored in a manner so as to ensure that no
significant growth or death of the test organism occurs before the dishes or plates
are used and that the surface of the agar layer is dry at the time of use.
Using the
appropriate buffer solutions indicated in Tables 10.2 and 10.3, prepare
solutions of known concentration of the Standard Preparation and solutions of
the corresponding assumed concen-trations of the antibiotic to be examined.
Where directions have been given in the individual monograph for preparing the
solutions, these should be followed and further dilutions made with buffer
solution as indicated in Table 10.3. Apply the solutions to the surface of the
solid medium in sterile cylinders or in cavities prepared in the agar. The
volume of soluiton added to each cylinder or cavity must be uniform and
sufficient almost to fill the holes when these are used. When paper discs are
used these should be sterilised by exposure of both sides under a sterilising
lamp and then impregnated with the standard solutions or the test solutions and
placed on the surface of the medium. When Petri dishes are used, arrange the
solutions of the Standard Preparation and the antibiotic to be examined on each
dish so that they alternate around the dish and so that the highest
concentrations of standard and test preparations are not adjacent. When plates
are used, place the solutions in a Latin square design, if the plate is a
square, or if it is not, in a randomised block design. The same random design
should not be used repeatedly.
Leave the
dishes or plates standing for 1 to 4 hours at room temperature or at 4°, as
appropriate, as a period of pre-incubation diffusion to minimise the effects of
variation in time between the applica-tion of the different solutions. Incubate
them for about 18 hours at the temperature indicated in Table 10.3. Accurately
measure the diameters or areas of the circular inhibition zones and calculate
the results.
Selection
of the assay design should be based on the requirements stated in the
individual mono-graph. Some of the usual assay designs are as follows.
[A.1] One-Level Assay with Standard Curve
Standard solution. Dissolve
an accurately weighted quantity of the Standard Preparation of the antibiotic, previously dried where
necessary, in the solvent specified in Table 10.3, and then dilute to the
required concentration, as indicated, to give the stock solution. Store in a
refrigerator and use within the period indicated. On the day of the assay,
prepare from the stock solutions, 5 dilutions (solutions S1 to S5)
representing five test levels of the standard and increasing stepwise in the
ratio of 4 : 5. Use the dilution specified in Table 10.3 and a sequence such
that the middle or median has the concentration given in the table.
Sample solution. From the information available
for the antibiotic preparation which is being examined (the “unknown”) assign to it an assumed potency per unit
weight or volume and on this assumption prepare on the day of the assay a stock
solution with the same solvent as used for the standard. Prepare from this
stock solution a dilution to a concentration equal to the median level of the standard
to give the sample solution.
Method. For preparing the standard curve,
use a total of 12 Petri dishes or plates to accommo-date 72 cylinders or
cavities. A set of three plates (18 cylinders or cavities) is used for each
dilution. On each of the three plates of a set fill alternate cylinders or
cavities with solution S3 (representing the median concentration of
the standard solution) and each of the remaining 9 cylinders or cavities with
one of the other 4 dilutions of the standard solution. Repeat the process for
the other 3 dilutions of the standard solutions. For each unknown preparation
use a set of three plates (18 cylinders or cavities) and fill alternate
cylinders or cavities with the sample solution and each of the remaining 9
cylinders of cavities with solution S3.
Incubate
the plates for about 18 hours at the specified temperature and measure the
diameters or the zones of inhibition.
Estimation of potency. Average
the readings of solution S3 and
the readings of the concentra-tion tested on each set of three plates, and
average also all 36 readings of solution S3. The average of the 36
readings of soluiton S3 is the correction point for the curve.
Correct the average value obtained for each concentration (S1, S2,
S4 and S5) to the figure it would be if the readings for
solution S3 for that set of three plates were the same as the
correction point. Thus, in correcting the value obtained with any
concentration, say S1, if the average of 36 readings of S3
is, for example, 18.0 mm and the average of the S3 concentrations on
one set of three plates is 17.8 mm, the correction is + 0.2 mm. If the average
reading of S1 is 16.0 mm, the corrected reading of S1 is
16.2 mm. Plot these corrected values including the average of the 36 readings
for solutions S3 on two-cycle semilog paper, using the
concentrations in Units or μg per ml
(as the ordinate logarithmic scale) and the diameter of the zones of inhibition
as the abscissa. Draw the straight response line either through these points by
inspection or through the points plotted for highest and lowest zone diameters
obtained by means of the following expressions :
L =
[ 3a + 2b + c – e] / 5 ; H = [ 3e + 2d + c – a]
/5
where
L = the
calculated zone diameter for the lowest concentration of the standard curve
response line.
H = the
calculated zone diameter for the highest concentration of the standard curve
response line.
c = average zone diameter of 36
readings of the reference point standard solution.
a, b, d, e = corrected average values for the
other standard solutions, lowest to highest
concentrations, respectively.
Average
the zone diameters for the sample solution and for solutions S3 on
the plates used for the sample soluiton. If the sample gives a large average
zone size than the average of the standard (solution S3), add the
difference between them to the zone size of solution S 3 of the
standard response line. If the average sample zone size is smaller than the
standard values, subtract the difference between them from the zone size of
solution S3 of the standard response line. From the response line
read the concentration corresponding to these corrected values of zone sizes.
From the dilution factors the potency of the sample may be calculated.
[A.2] Two-Level Factorial Assay
Prepare
parallel dilutions containing 2 levels of both the standard (S1 and
S2) and the unkown (U1 and U2). On each of
four or more plates, fill each of its four cylinders or cavities with a
different test dilution, alternating standard and unknown. Keep the plates at
room temperature and measure the diam-eters of the zones of inhibition.
Estimation of potency. Sum the
diameters of the zones of each dilution and calculate the % potency of the sample (in terms of the standard) from the
following equation :
% potency
= Antilog (2.0 + a log I)
wherein a may have a positive or negative value
and should be used algebracially and
where a = (U1 + U2 ) –
(S1 + S2 ) / (U1 + U2) + (S1
– S2)
U1
and U2 are the sums of the zone diameters with solutions of the
unknown of high and low levels.
S1
and S2 are the sums of the zone diameters with solutions of the
standard of high and low levels.
I = ratio
of dilutions.
If the
potency of the sample is lower than 60% or greater than 150% of the standard,
the assay is invalid and should be repeated using higher or lower dilutions of
the same solutions. The potency of the sample may be calculated from the
expression.
[ % potency
× assumed potency of the sample ] / 100
[A.3] Other Designs
(1) Factorial
assay containing parallel dilution of three test levels of standard and the
unknown.
(2) Factorial
assay using two test levels of standard and two test levels of two different
un- knowns.
[B] Turbidimetric or Tube Assay Method
The
method has the advantage of a shorter
incubation period for the growth of the test organism (usually 3 to 4
hours) but the presence of solvent residues or other inhibitory substances
affects this assay more than the cylinder-plate assay and care should be taken
to ensure freedom from such sub-stances in the final test solutions. This
method is not recommended for cloudy or turbid preparations.
Prepare
five different concentrations of the standard solution for preparing the
standard curve by diluting the stock solution of the Standard Preparation of
the antibiotic (Table 10.3) and increasing stepwise in the ratio 4 : 5. Select
the median concentration (Table 10.3) and dilute the solution of the substance
being examined (unknown) to obtain approximately this concentration. Place 1 mL
of each concentration of the standard solution and of the sample solution in
each of the tubes in duplicate. To each tube add 9 ml of nutrient medium (Table
10.3) previously seeded with the appropriate test organ-ism (Table 10.3).
At the
same time prepare three control tubes, one containing the inoculated culture
medium (cul-ture control), another identical with it but treated immediately
with 0.5 mL of dilute formaldehyde
solu-tion (blank) and a third containing uninoculated culture medium.
Place all
the tubes, randomly distributed or in a randomized block arrangement, in an
incubator or a water-bath and maintain them at the specified temperature (Table
10.3) for 3 to 4 hours. After incubation add 0.5 mL of dilute formaldehyde solution to each tube. Measure the growth of
the test organism by determining the absorbance
at about 530 nm of each of the solutions in the tubes against the blank.
Estimation of potency. Plot the
average absorbances for each concentration of the standard on semi-logarithmic
paper with the absorbances on the arithmetic scale and concentrations on
the logarith-mic scale. Construct the best straight response line through the
points either by inspection or by means of the following expressions :
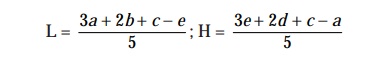
where
L = the
calculated absorbance for the lowest concentration of the standard response
line.
H = the
calculated absorbance for the highest concentration of the standard response
line.
a, b, c, d, e = average absorbance values for
each concentration of the standard response line lowest to highest
respectively.
Plot the
values obtained for L and H and connect the points. Average the absorbances for
the sample and read the antibiotic concentration from the standard response
line. Multiply the concentration by the appropriate dilution factors to obtain
the antibiotic content of the sample.
Precision of Microbiological Assays
The
fiducial limits of error of the estimated potency should be not less than 95%
and not more than 105% of the estimated potency unless otherwise stated in the
individual monograph. This degree of precision is the minimum acceptable for
determining that the final product complies with the official requirements and
may be inadequate for those deciding, for example, the potency which should be
stated on the label or used as the basis for calculating the quantity of an
antibiotic to be incorporated in a preparation. In such circumstances, assays
of greater precision may be desirable with, for instance, fiducial limits of
error of the order of 98% to 102%. With this degree of precision, the lower
fiducial limit lies close to the estimated potency. By using this limit,
instead of the estimated potency, to assign a potency to the antibiotic either
for labelling or for calculating the quantity to be included in a prepara tion,
there is less likelihood of the final preparation subsequently failing to comply
with the official requirements for potency.
Related Topics
