Antifungal Therapy
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Fungi
The choice and dose of an antifungal will depend upon the nature of the condition, whether there are any underlying diseases, the health of the patient and whether antifungal resistance has been identified as compromising therapy.
ANTIFUNGAL THERAPY
The choice and dose of
an antifungal will depend upon the nature of the condition, whether there are
any underlying diseases, the health of the patient and whether antifungal
resistance has been identified as compromising therapy. Part of the difficulty
in designing effective antifungal agents lies in the fact that fungi are
eukaryotic organisms so agents that will kill fungi may also have a deleterious
effect on human tissue. The ideal antifungal drug should target a pathway or
process specific to the fungal cell, so reducing the possibility of damaging
tissue and inducing unwanted side effects.
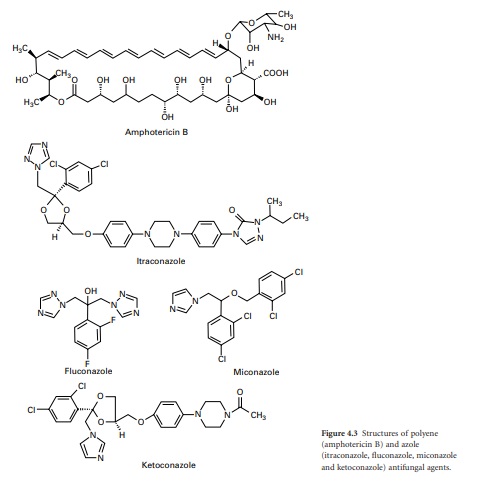
Polyene antifungals
Polyene antifungals are
characterized by having a large macrolide ring of carbon atoms closed by the
formation of an internal ester or lactone (Figure 4.3). In addition, polyenes have
a large number of hydroxyl groups distributed along the macrolide ring on
alternate carbon atoms. This combination of highly polar and nonpolar regions
within the molecule renders the polyenes amphiphatic, i.e. having hydrophobic
and hydrophilic regions in the one molecule, which assists solubility in lipid
membranes.
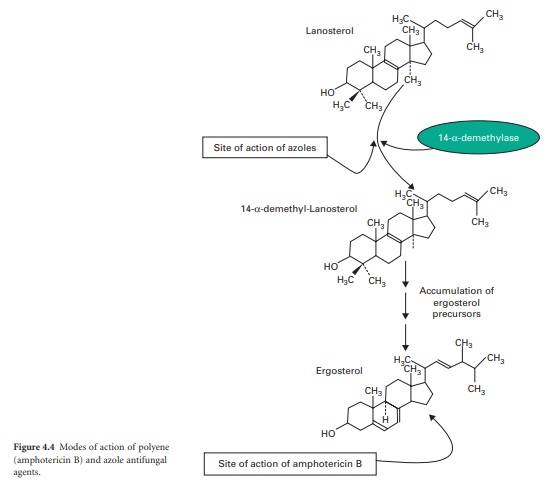
The principal polyenes are
amphotericin B and nystatin. Amphotericin B is produced by the bacterium Streptomyces nodosus and its activity is
due to the ability to bind
ergosterol, which is the dominant sterol in fungal cell membranes, and
consequently increases membrane permeability by the formation of pores (Figure
4.4). The action of amphotericin B seems to rely on the formation of pores
through which intracellular contents can escape from the cell. Amphotericin B
can lead to renal damage during prolonged antifungal therapy. Amphotericin B is
active against a broad range of fungal pathogens and is considered the ‘gold
standard’ against which the activity of other antifungal agents is measured.
Because of its renal toxicity amphotericin B tends to be reserved for severe
cases of systemic fungal disease but recent formulations in which the drug is
encapsulated within liposomes have been shown to have reduced toxicity.
Nystatin was discovered
in 1950 and exhibits the same mode of action as amphotericin B but tends to
have lower solubility, which has restricted its use to the treatment of topical
infections. Although nystatin was effective for the treatment of conditions
such as oral and vaginal candidosis, its use has been overtaken by the introduction
of azole antifungal drugs.
Azole antifungals
The first generation of
azole antifungals revolutionized the treatment of mucosal and invasive fungal
infections, and azoles are still the most widely used group of antifungal
agents. The azole derivatives are classified as imidazoles or triazoles on the
basis of whether they have two or three nitrogen atoms in the fivemembered
azole ring (Figure 4.3). The azoles in current clinical use are clotrimazole,
miconazole, econazole and ketoconazole; newer drugs such as itraconazole,
fluconazole and voriconazole have important applications in the treatment of
systemic infections. Azoles function by interfering with ergosterol
biosynthesis by binding to the cytochrome P450 mediated enzyme known as 14αdemethylase (P450DM). This blocks the formation
of ergosterol by preventing the methylation of lanosterol (a precursor of ergosterol)
(Figure 4.4). This result in a reduction in the amount of ergosterol in the
fungal cell membrane which leads to membrane instability, growth inhibition and
cell death. An additional consequence of the block in ergosterol biosynthesis
is the build up of toxic intermediates which can prove fatal to the fungal
cell.
Azoles exhibit a broad
spectrum of activity in vitro, being
capable of inhibiting the growth of most Candida,
Cryptococcus and Aspergillus species, and dermatophytes. Miconazole was the first azole used to treat systemic fungal
infections but demonstrated a number of toxic side effects. Ketoconazole
produced high serum concentrations upon oral administration but had poor activity
against aspergillosis. In addition, ketoconazole was associated with a range of
side effects which limited its applicability. Newer triazoles such as
fluconazole and itraconazole have increased the options for dealing with fungal
infections. Fluconazole was introduced for clinical use in 1990, is water
soluble and shows good penetration and deposition into the pulmonary tissues; it
also reaches high levels in the cerebrospinal fluid and the peritoneal fluids.
Fluconazole has proved highly effective in the treatment of infections caused
by C. albicans but shows limited
activity against Aspergillus.
Itraconazole became available for clinical use in the late 1980s and was the
first azole with proven efficacy against Aspergillus.
Itraconazole is effective in treating severe Aspergillus infections and exhibits both fungicidal and fungistatic
effects. Upon ingestion itraconazole undergoes extensive hepatic metabolism
which yields up to 30 metabolites, a number of which retain antifungal
activity. Itraconazole is currently available as an intravenous formulation and
is widely used for the treatment of severe Aspergillus
infection in this form. Fluconazole and itraconazole demonstrate significantly
reduced side effects compared to ketoconazole. Novel azole drugs with increased
ability to inhibit the fungal 14α demethylase are also becoming available. These agents, which
include voriconazole, posaconazole and ravuconazole, have a wider spectrum of
activity than fluconazole and it has been suggested that some of them show
fungicidal effects to some species (e.g. Aspergillus
spp.). Voriconazole is one of the newest secondgeneration triazole antifungal
drugs and it shows good activity against pulmonary aspergillosis and cerebral
aspergillosis.
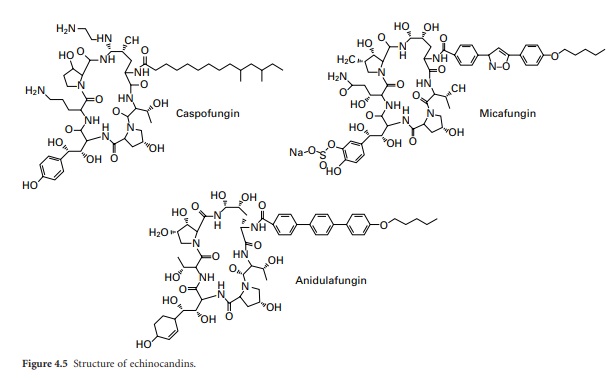
Echinocandins
The echinocandins are a
relatively new group of antifungal drug and are semisynthetic lipopeptides
comprising a cyclic hexapeptide core connected to a lateral fatty acid chain.
Three compounds of this group are currently in use: caspo-fungin, micafungin
and anidulafungin (Figure 4.5 ). Unlike conventional antifungal therapy that
targets ergosterol or its synthesis, the echinocandins target the synthesis of β-1-,3-glucan, the major polymer of the fungal
cell wall. The cell wall is essential to the fungus as it provides physical
protection, maintains osmotic stability, regulates cell shape, acts as a
scaffold for proteins, mediates cell–cell communication and is the site of a
number of enzymatic reactions. Inhibition of β-1-,3-glucan synthesis disrupts the structure of the growing cell
wall, resulting in osmotic instability and ballooning out of the intracellular
contents as a result of high osmotic pressure, and ultimately ends in cell lysis.
Caspofungin has
demonstrated in vitro antifungal
activity against various filamentous fungi and yeasts. It has activity against
different Aspergillus species
including A. fumigatus, A. flavus, A. niger and A. terreus but
is considered to be more fungistatic than fungicidal. Conversely, caspofungin
is particularly fungicidal against a range of Candida species including species that are resistant (e.g. C. krusei) or isolates that are less
susceptible (e.g. C. dubliniensis, C. glabrata) to azoles, or resistant to
amphotericin B.
The fungal cell wall
represents an attractive target and the echinocandins have proven to offer a
safer alternative to conventional antifungal therapies. (i.e. polyenes and
azoles). Echinocandins display an unique mode of action which results in
defects in cell wall morphology and osmotic instability. As the cell wall is an
essential component for stability and ultimately virulence, the targeting of
the wall by echinocandins results in the efficient destruction of the fungal
cell. To avoid future problems with resistance, researches need to clarify the
precise interactions of the echinocandins with the target enzyme, and fully
examine the cell’s complex response to this agent.
Synthetic antifungal agents
Flucytosine is a synthetic
fluorinated pyrimidine which has been used as an oral antifungal agent and
demonstrates good activity against a range of yeast species and moderate levels
of activity against Aspergillus
species. Two modes of action have been proposed for flucytosine. One involves
the disruption of protein synthesis by the inhibition of DNA synthesis while
the other possible mode of action is the depletion in the amino acid pools
within the cell as a result of inhibition of protein synthesis. In general
yeast cells increase in size when exposed to levels of flucytosine lower than
the minimum inhibitory concentration (MIC) and display alterations in their
surface morphology, both of which can be interpreted as a result of an
imbalance in the control of cellular growth. Many fungi are inherently
resistant to flucytosine or develop resistance after a relatively short
exposure and resistance has been attributed to alteration in the enzyme
(cytosine deaminase) required to process flucytosine once inside the cell or to
an elevation in the amount of pyrimidine synthesis. The problem of resistance
has limited the use of flucytosine so that now it is generally used in
combination with an antifungal agent (e.g. amphotericin B) where it can
potentiate the effect of the second agent.
Related Topics
