Chapter Summary, Questions Answers - Blood clotting
| Home | | Biochemistry |Chapter: Biochemistry : Blood clotting
Blood clotting (coagulation) is designed to rapidly stop bleeding from a damaged blood vessel in order to maintain a constant blood volume (hemostasis).
CHAPTER SUMMARY
Blood clotting
(coagulation) is designed to rapidly stop bleeding from a damaged blood vessel
in order to maintain a constant blood volume (hemostasis). Coagulation is accomplished
through formation of a clot (thrombus) consisting of a plug of platelets
(thrombocytes) and a meshwork of the protein fibrin (Figure 34.25). Wound to a
tissue damages blood vessels and exposes collagen. Platelets adhere to the
exposed collagen, get activated, and aggregate to form a platelet plug.
Adhesion is mediated by von Willebrand Factor (VWF). VWF binds collagen, and
platelets bind VWF via GPIb within a receptor complex on the platelet surface.
Deficiency of VWF results in von Willebrand disease, the most common inherited
coagulopathy. Once adhered, platelets get activated. Platelet activation
involves changes in shape (discoidal to spherical with pseudopodia) and
degranulation, the process by which platelets release the contents of their storage
granules. Thrombin is the most potent activator of platelets. Thrombin binds to
protease-activated G protein–coupled receptors on the surface of platelets.
Activated platelets release substances that cause vasoconstriction (serotonin
and thromboxane A2 [TXA2] ) , recruit and activate other
platelets (adenosine diphosphate and TXA2) and support the formation
of a fibrin clot (factor [F] V, FXIII, and fibrinogen). Activation causes
changes in platelets that lead to their aggregation. Structural changes in a
surface receptor (GPIIb/IIIa) expose binding sites for fibrinogen. Fibrinogen
molecules link activated platelets to one another. The fibrinogen is activated
to fibrin by thrombin and then cross-linked by FXIIIa, a transglutaminase
coming both from the blood and from platelets. The initial loose plug of
platelets (primary hemostasis) is strengthened by the fibrin meshwork
(secondary hemostasis).
The formation of the
fibrin meshwork involves the extrinsic and intrinsic pathways (and their
associated protein factors) that converge at FXa to form the common pathway.
Many of the protein factors are serine proteases with trypsin-like specificity.
Ca 2+ binds the negatively charged γ-carboxyglutamate (Gla) residues present in
certain of the clotting proteins (FII, FVII, FIX, and FX), facilitating the
binding of these proteins to exposed phosphatidylserine at the site of injury
and on the surface of platelets. γ-Glutamyl carboxylase and its coenzyme, the
hydroquinone form of vitamin K, are required for formation of Gla residues. In
the reaction, vitamin K gets oxidized to the nonfunctional epoxide form.
Warfarin, a synthetic analog of vitamin K used clinically to reduce clotting,
inhibits the enzyme vitamin K epoxide reductase that regenerates the functional
reduced form. The extrinsic pathway is initiated by exposure of FIII (tissue
factor [TF]), an accessory protein, in vascular subendothelium. Exposed TF
binds a circulating Gla-containing protein, FVII, activating it through
conformational change. The TF–FVIIa complex then binds and activates FX by
proteolysis. The extrinsic pathway is rapidly inhibited by tissue factor
pathway inhibitor. The intrinsic pathway is initiated by FXIIa. FXIIa activates
FXI, and FXIa activates FIX. FIXa combines with FVIIIa (an accessory protein),
and the complex activates FX. FVIII deficiency results in hemophilia A, whereas
FIX deficiency results in the less common hemophilia B. FXa associates with FVa
(an accessory protein), forming prothrombinase that cleaves prothrombin (FII) t
o thrombin (FIIa) . Thrombin then cleaves fibrinogen to fibrin (FIa). Fibrin
monomers associate, forming a soluble (soft) fibrin clot. The fibrin molecules
get cross-linked by FXIIIa, forming an insoluble (hard) fibrin clot. Proteins
synthesized by the liver and by blood vessels themselves balance coagulation
with anticoagulation. Antithrombin III, a serine protease inhibitor, or serpin,
binds to and removes thrombin from the blood. Its affinity for thrombin is
increased by heparin, which is used therapeutically to limit clot formation.
Protein C, a Gla-containing protein, is activated by the
thrombin–thrombomodulin complex. Thrombomodulin decreases thrombin’s affinity
for fibrinogen, converting it from a protein of coagulation to a protein of
anticoagulation. Protein C in complex with protein S (a Gla-containing protein)
forms the activated protein C (APC) complex that cleaves the accessory proteins
FVa and FVIIIa. Factor V Leiden is resistant to APC. It is the most common
inherited thrombophilic condition in the United States. The fibrin clot is
cleaved (fibrinolysis) by the protein plasmin, a serine protease that is
generated from plasminogen by plasminogen activators such as tissue plasminogen
activator (TPA, t-PA). Recombinant TPA is used clinically. Disorders of
platelets and coagulation proteins can result in deviations in the ability to
clot. Prothrombin time and activated partial thromboplastin time are used to
evalulate the clotting cascade.
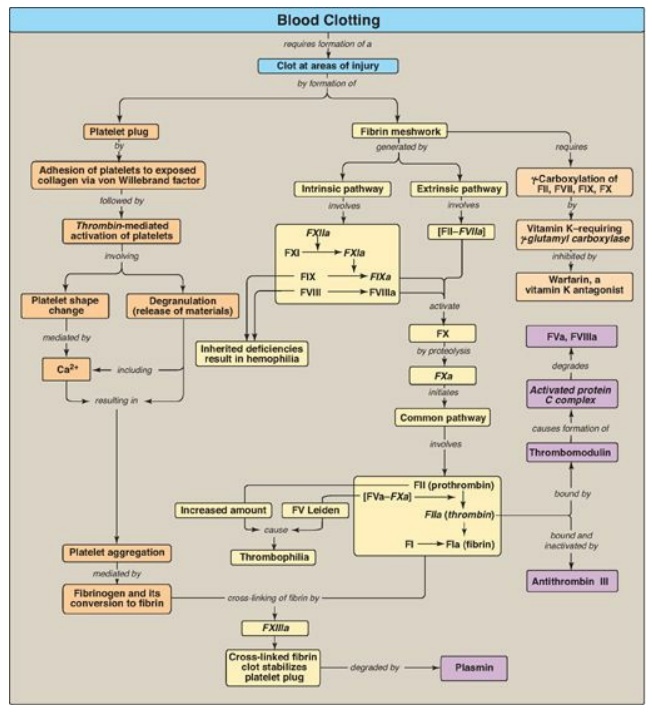
Figure 34.25 Key concept map
for blood clotting. a = active; F = factor.
Study Questions
Choose the ONE best answer.
For Questions
31.1–31.5, match the most appropriate protein of clotting to the description.
A. FI
B. FII
C. FIII
D. FV
E. FVII
F. FVIII
G. FIX
H. FX
I. FXI
J. FXIII
34.1 This factor
activates components of the intrinsic, extrinsic, and common pathways.
34.2 This factor
converts the soluble clot to an insoluble clot.
34.3 This factor
initiates the common pathway.
34.4 This factor is an
accessory protein that potentiates the activity of factor Xa.
34.5 This factor is a
γ-carboxyglutamate–containing serine protease of the extrinsic pathway.
Correct answers = B, J, H, D, E. Thrombin (FII) is formed in the
common pathway and activates components in each of the three pathways of the
clotting cascade. Factor (F)XIII, a transglutaminase, covalently cross-links
associated fibrin monomers, thereby converting a soluble clot to an insoluble
one. The generation of FXa by the intrinsic and extrinsic pathways initiates
the common pathway. FV increases the activity of FXa. It is one of three
accessory (nonprotease) proteins. The others are FIII (tissue factor) and FVIII
(complexes with FIX to activate FX). FVII is a γ-carboxyglutamate–containing
serine protease that complexes with FIII in the extrinsic pathway.
34.6 In which patient would prothrombin time be
unaffected and activated partial thromboplastin time be prolonged?
A. A patient on aspirin
therapy
B. A patient with
end-stage liver disease
C. A patient with hemophilia
D. A patient with
thrombocytopenia
Correct answer = C. Prothrombin time (PT) measures the
activity of the extrinsic through the common pathways, and activated partial
thromboplastin time (aPTT) measures the activity of the intrinsic through the
common pathways. Patients with hemophilia are deficient in either factor
(F)VIII (hemophilia A) or FIX (hemophilia B), components of the common pathway.
They have an intact extrinsic pathway. Therefore, the PT is unaffected, and the
aPTT is prolonged. Patients on aspirin therapy and those with thrombocytopenia
have alterations in platelet function and number, respectively, and not in the
proteins of the clotting cascade. Therefore, both the PT and the aPTT are
unaffected. Patients with end-stage liver disease have decreased ability to
synthesize clotting proteins. They show prolonged PT and aPTT.
34.7 Which one of the following can be ruled out in
a patient with thrombophilia?
A. A deficiency of
antithrombin III
B. A deficiency of factor IX
C. A deficiency of protein
C
D. An excess of
prothrombin
E. Expression of factor
V Leiden
Correct answer = B. Symptomatic deficiencies in
clotting factors will present with a decreased ability to clot (coagulopathy).
Thrombophilia, however, is characterized by an increased tendency to clot.
Choices A, C, D, and E result in thrombophilia.
34.8 Current guidelines for the treatment of
patients with acute ischemic stroke (a stroke caused by a blood clot
obstructing a vessel that supplies blood to the brain) include the recommendation
that tissue plasminogen activator (TPA) be used shortly after the onset of
symptoms. The basis of the recommendation for TPA is that it activates:
A. antithrombin III.
B. the activated
protein C complex.
C. the receptor for von
Willebrand factor.
D. the serine protease
that degrades fibrin.
E. thrombomodulin.
Correct answer = D. Tissue plasminogen activator (TPA)
converts plasminogen to plasmin. Plasmin (a serine protease) degrades the
fibrin meshwork, removing the obstruction to blood flow. Antithrombin III in
association with heparin binds thrombin and carries it to the liver, decreasing
thrombin’s availability in the blood. The activated protein C complex degrades
the accessory proteins FV and FVIII. The platelet receptor for von Willebrand
factor is not affected by TPA. Thrombomodulin binds thrombin and converts it
from a protein of coagulation to one of anticoagulation by decreasing its
activation of fibrinogen and increasing its activation of protein C.
34.9 The adhesion, activation, and aggregation of
platelets provide the initial plug at the site of vessel injury. Which of the
following statements concerning the formation of this platelet plug is correct?
A. Activated platelets
undergo a shape change that decreases their surface area.
B. Formation of a
platelet plug is prevented in intact vessels by the production of thromboxane
A2 by endothelial cells.
C. The activation phase
requires production of cyclic adenosine monophosphate.
D. The adhesion phase is mediated by the binding of
platelets to von Willebrand factor via glycoprotein Ib.
E. Thrombin activates
platelets by binding to a protease-activated G protein– coupled receptor and
causing activation of protein kinase A.
Correct answer = D. The adhesion phase of platelet plug
formation is initiated by the binding of von Willebrand factor to a receptor
(glycoprotein Ib) on the surface of platelets. Shape change from discoidal to
spherical with pseudopodia increases the surface area of platelets. Thromboxane
A2 is made by platelets. It causes platelet activation and vasoconstriction.
Adenosine diphosphate is released from activated platelets, and it itself
activates platelets. Thrombin works primarily through receptors coupled to Gq
proteins causing activation of phospholipase C.
34.10 Several days after having had their home
treated for an infestation of rats, the parents of a 3-year-old girl become
concerned that she might be ingesting the poison-containing pellets. After
calling the Poison Hotline, they take her to the Emergency Department. Blood studies
reveal a prolonged prothrombin and activated partial thromboplastin time and a
decreased concentration of factor (F)II, FVII, FIX, and FX. Why might
administration of vitamin K be a rational approach to the treatment of this
patient?
Many rodent poisons are
super warfarins, drugs that have a long half-life in the body. Warfarin
inhibits γ-carboxylation (production of γ-carboxyglutamate, or Gla, residues),
and the clotting proteins reported as decreased are the Gla-containing
proteases of the clotting cascade. [Note: Proteins C and S of anticlotting are
also Gla-containing proteins.] Because warfarin functions as a vitamin K
antagonist, administration of vitamin K is a rational approach to treatment.
Related Topics
