Fibrin Meshwork Formation
| Home | | Biochemistry |Chapter: Biochemistry : Blood clotting
The formation of the fibrin meshwork involves two unique pathways that converge to form a common pathway.
FIBRIN MESHWORK FORMATION
The formation of the
fibrin meshwork involves two unique pathways that converge to form a common
pathway (Figure 34.2). In each pathway, the major components are proteins
(called factors) designated by Roman numerals. The factors are glycoproteins
that are synthesized and secreted by the liver, primarily. [Note: Several
factors are also denoted by alternative names. For example, active factor X
(FXa), the point of pathway convergence, is also known as Stuart factor.]
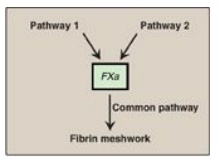
Figure 34.2 Three pathways
involved in formation of the fibrin meshwork.
A. Protolytic cascade
Within the pathways, a
cascade is set up in which proteins are converted from an inactive form, or
zymogen, to an active form by proteolytic cleavage in which the protein product
of one activation reaction initiates another. The active form of a factor is
denoted by a lower case “a” after the numeral. The active proteins FIIa, FVIIa,
FIXa, FXa, FXIa, and FXIIa are enzymes that function as serine proteases with
trypsin-like specificity and, therefore, cleave a peptide bond on the carboxyl
side of an arginine or lysine residue in a polypeptide. For example, FIX
(Christmas factor) is activated through cleavage at arginine 145 and arginine
180 by FXIa (Figure 34.3). The proteolytic cascade results in enormous rate
acceleration, because one active protease can produce many molecules of active
product each of which, in turn, can activate many molecules of the next protein
in the cascade. In some cases, activation can be caused by a conformational
change in the protein in the absence of proteolysis. [Note: Nonproteolytic
proteins play a role as accessory proteins (cofactors) in the pathways. FIII,
FV, and FVIII are the accessory proteins.]
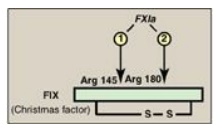
Figure 34.3 Activation of FIX
(Christmas factor) via proteolysis by the serine protease FXIa. [Note:
Activation can occur by conformational change for some of the factors.] a =
active; Arg = arginine.
B. Role of phosphatidylserine and calcium
The presence of the
negatively charged phospholipid phosphatidylserine (PS) and positively charged
calcium ions (Ca2+) accelerates the rate of some steps in the
cascade.
1. Negatively charged phosphatidylserine: PS is located primarily on the
intracellular (cytosolic) face of the plasma membrane. Its exposure signals
injury to the endothelial cells that line blood vessels. PS is also exposed on
the surface of activated platelets.
2. Calcium ions: Ca2+ binds the negatively charged
γ-carboxyglutamate (Gla) residues present in certain clotting serine proteases
(FII, FVII, FIX, and FX), facilitating the binding of these proteins to exposed
phospholipids (Figure 34.4). The Gla residues are good chelators of Ca2+
because of their two adjacent negatively charged carboxylate groups (Figure
34.5). [Note: The use of chelating agents such as sodium citrate to bind Ca2+
in blood-collecting tubes or bags prevents the blood from clotting.]
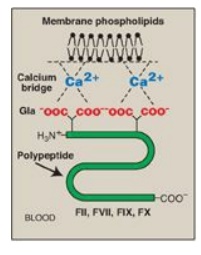
Figure 34.4 Ca2+
facilitates the binding of γ-carboxyglutamate (Gla)- containing factors to
membrane phospholipids. F = factor.
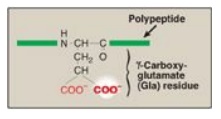
Figure 34.5 Gla residue.
C. Formation of γ-carboxyglutamate residues
γ-Carboxylation is a
posttranslational modification in which 9–12 glutamate residues (at the amino or
N terminus of the target protein) get carboxylated at the γ carbon, thereby
forming γ-carboxyglutamate (Gla) residues. The process occurs in the rough
endoplasmic reticulum (RER) of the liver.
1. γ-Carboxylation: This carboxylation reaction
requires a protein substrate, O2, CO2, γ-glutamyl
carboxylase, and the hydroquinone form of vitamin K as a coenzyme (Figure
34.6). In the reaction, the hydroquinone form of vitamin K gets oxidized to its
epoxide form as O2 is reduced to water. [Note: Vitamin K, a fat-soluble
vitamin, is reduced from the quinone form to the hydroquinone coenzyme form by
vitamin K reductase (Figure 34.7).]
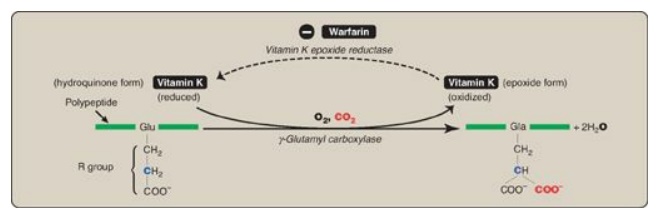
Figure 34.6 γ-Carboxylation of a glutamate (Glu) residue to γ-carboxyglutamate (Gla) by vitamin K-requiring γ-glutamyl carboxylase. The γ carbon is shown in blue.
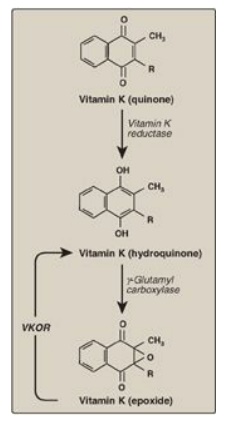
Figure 34.7 The vitamin K cycle. VKOR = vitamin K epoxide reductase.
2. Inhibition by warfarin: The formation of Gla residues is
sensitive to inhibition by warfarin, a synthetic analog of vitamin K that
inhibits the enzyme vitamin K epoxide reductase (VKOR). The reductase, an
integral protein complex of the RER membrane, is required to regenerate the
functional hydroquinone form of vitamin K from the epoxide form generated in
the γ-carboxylation reaction. Thus, warfarin is an anticoagulant that inhibits
clotting by functioning as a vitamin K antagonist. Warfarin salts are used
therapeutically to limit clot formation. [Note: Warfarin is used commercially
as a pest-control agent such as in rat poison. It was developed by the
Wisconsin Alumni Research Foundation, hence the name.]
Genetic differences (genotypes) in the gene for
subunit 1 of the VKOR complex (VKORC1) influence patient response to warfarin.
For example, a polymorphism in the promoter region of the gene decreases gene
expression, resulting in less VKOR being made, thereby necessitating a lower
dose of warfarin to achieve a therapeutic level. Polymorphisms in the cytochrome
P450 enzyme (CYP2C9) that metabolizes warfarin are also known. In 2010, the
U.S. Food and Drug Administration added a genotype-based dose table to the
warfarin label (package insert). The influence of genetics on an individual’s
response to drugs is known as pharmacogenetics.
D. Pathways
Three distinct pathways
are involved in formation of the fibrin meshwork: the extrinsic pathway, the
intrinsic pathway, and the common pathway. Production of FXa by the extrinsic
and intrinsic pathways initiates the common pathway (see Figure 34.2).
1. Extrinsic pathway: This pathway involves a protein, tissue factor (TF), that is not in the blood but becomes exposed when blood vessels get injured. TF (FIII) is a transmembrane glycoprotein abundant in vascular subendothelium. It is an extravascular accessory protein and not a protease. Any injury that exposes FIII to blood rapidly (within seconds) initiates the extrinsic (or TF) pathway. Once exposed, TF binds a circulating Gla-containing protein, FVII, activating it through conformational change. [Note: FVII can also be activated proteolytically by thrombin (see Section 3. below).] Binding of FVII to TF requires the presence of Ca2+ and phospholipids. The TF–FVIIa complex then binds and activates FX by proteolysis (Figure 34.8). Therefore, activation of FX by the extrinsic pathway occurs in association with the membrane. The extrinsic pathway is quickly inactivated by tissue factor pathway inhibitor (TFPI) that, in a FXa-dependent process, binds to the TF–FVIIa complex and prevents further production of FXa. [Note: TF and FVII are unique to the extrinsic pathway.]
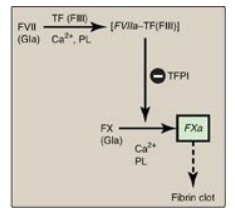
Figure 34.8 The extrinsic or
tissue factor (TF) pathway. Binding of FVII to exposed TF (FIII) activates
FVII. [Note: The pathway is quickly inhibited by tissue factor pathway
inhibitor (TFPI).] F = factor; Gla = γ-carboxyglutamate; PL = phospholipid; a =
active.
2. Intrinsic pathway: All of the protein factors involved
in the intrinsic pathway are present in the blood and are, therefore,
intravascular. The intrinsic pathway involves two phases: the contact phase and
the FX-activation phase, each with known deficiencies.
a. Contact phase: This phase results in the
activation of FXII (Hageman factor) to FXIIa by conformational change through
binding to a negative surface. Deficiencies in FXII (or in the other proteins
of this phase, high-molecular-weight kininogen and prekallikrein) do not result
in bleeding problems, calling into question the importance of this phase in
coagulation. However, the contact phase does play a role in inflammation.
[Note: FXII can be activated proteolytically by thrombin (see Section 3.
below)].
b. Factor X–activation phase: The sequence of events leading to
the activation of Factor X to FXa by the intrinsic pathway is initiated by
FXIIa (Figure 34.9). FXIIa activates FXI, and FXIa activates FIX, a
Gla-containing protein. FIXa combines with FVIIIa (a bloodborne accessory
[nonenzymatic] protein), and the complex activates FX, a Gla-containing serine
protease. [Note: The complex containing FIXa, FVIIIa, and FX forms on exposed
negatively charged membrane regions, and FX gets activated to FXa. This complex
is sometimes referred to as Xase. Binding of the complex to membrane
phospholipids requires Ca2+.]
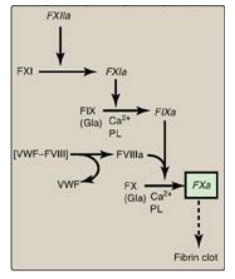
Figure 34.9 FX activation phase of the intrinsic pathway. [Note: von Willebrand factor (VWF) stabilizes FVIII in the circulation.] Gla = γ-carboxyglutamate; PL = phospholipid; a = active; F = factor.
c. Factor XII deficiency: A deficiency in FXII does not lead
to a bleeding disorder. This is because FXI, the next protein in the cascade,
can be activated proteolytically by thrombin (see Section 3. below).
d. Hemophilia: Hemophilia is a coagulopathy, a defect in the ability to clot. Hemophilia A (the most common form of hemophilia) results from deficiency of FVIII, whereas deficiency of FIX results in hemophilia B. Each deficiency is characterized by decreased and delayed ability to clot and/or formation of abnormally friable (easily disrupted) clots. This can be manifested, for example, by bleeding into the joints (Figure 34.10). The extent of the factor deficiency determines the severity of the disease. Current treatment for hemophilia is factor replacement therapy using factors obtained from pooled human blood or from recombinant DNA technology. Gene replacement therapy is a goal. Because the genes for both proteins are on the X chromosome, hemophilia is an X-linked disorder. [Note: Deficiency of FXI results in a bleeding disorder that sometimes is referred to as hemophilia C.]
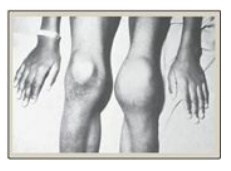
Figure 34.10 Acute bleeding into joint spaces (hemarthrosis) in an individual with hemophilia.
The inactivation of the extrinsic pathway by TFPI results in dependence on the intrinsic pathway for continued production of FXa. This explains why individuals with hemophilia bleed even though they have an intact extrinsic pathway.
3. Common pathway: FXa produced by both the intrinsic
and the extrinsic paths initiates the common pathway, a sequence of events that
results in the generation of fibrin (FIa) (Figure 34.11) . FXa associates with
FVa (a bloodborne accessory [nonenzymic] protein) and, in the presence of Ca2+
and phospholipids, forms a membrane-bound complex referred to as
prothrombinase. The complex cleaves prothrombin (FII) to thrombin (FIIa).
[Note: FVa potentiates the proteolytic activity of FXa.] The binding of Ca2+
to the Gla residues in FII facilitates the binding of FII to the membrane and
to the prothrombinase complex, with subsequent cleavage to thrombin. Cleavage
excises the Gla-containing region, releasing thrombin from the membrane and,
thereby, freeing it to activate fibrinogen (FI) in the blood. [Note: This is
the only example of cleavage of a Gla protein that results in the release of a
Gla-containing peptide. The peptide travels to the liver where it is thought to
act as a signal for increased production of clotting proteins.] Oral, direct
inhibitors of FXa have been approved for limited clinical use as
anticoagulants.
A common point mutation (G20210A) in which an
adenine (A) replaces a guanine (G) at nucleotide 20210 in the 3′ untranslated
region of the gene for prothrombin leads to increased levels of prothrombin in
the blood. This results in thrombophilia, a condition characterized by an increased
tendency to clot.
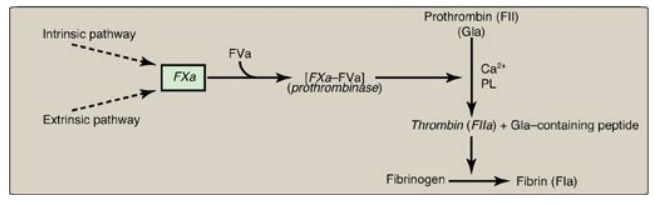
Figure 34.11 Generation of
fibrin by FXa and the common pathway. F = factor; Gla = γ-carboxyglutamate; PL
= phospholipid; a = active.
a. Conversion of fibrinogen to fibrin by thrombin: Fibrinogen is a soluble glycoprotein
made by the liver. It consists of dimers of three different polypeptide chains
[(Aα)2(Bβ)2(γ)2] held together at the N termini by disulfide bonds. [Note: Aα
and Bβ each represent a single polypeptide.] The N termini of the Aα and Bβ
chains form “tufts” on the central of three globular domains (Figure 34.12).
The tufts are negatively charged and result in repulsion between fibrinogen
molecules. Thrombin cleaves the charged tufts (releasing fibrinopeptides A and
B), and fibrinogen becomes fibrin. As a result of the loss of charge, the
fibrin monomers are able to noncovalently associate in a staggered array, and a
soft (soluble) fibrin clot is formed.
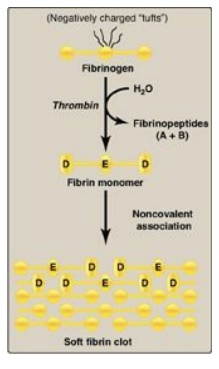
Figure 34.12 Conversion of
fibrinogen to fibrin and formation of the soft fibrin clot. [Note: D and E
refer to domains on fibrin.]
b. Cross-linking of fibrin: The associated linked. This
converts the soft clot to fibrin molecules get covalently cross-a hard
(insoluble) clot. FXIIIa, a transglutaminase, covalently links the γ-carboxamide
of a glutamine residue in one fibrin molecule to the ε-amino of a lysine
residue in another through formation of an isopeptide bond and release of
ammonia (Figure 34.13). [Note: FXIII is also activated by thrombin.]
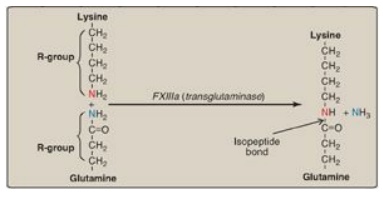
Figure 34.13 Cross-linking of fibrin. FXIIIa forms a covalent isopeptide bond between a lysine residue and a glutamine residue. F = factor.
c. Importance of thrombin: The activation of FX by the
extrinsic path provides the “spark” of FXa that results in the initial
activation of thrombin. Active thrombin then activates factors of the common
(FV, FI, FXIII), intrinsic (FXI, FVIII), and extrinsic (FVII) pathways (Figure
34.14). It also activates FXII of the contact phase. The extrinsic pathway,
then, initiates clotting by the generation of FXa, and the intrinsic pathway
amplifies and sustains clotting after the extrinsic pathway has been inhibited
by TFPI. [Note: Hirudin, a peptide secreted from the salivary gland of
medicinal leeches, is a potent, direct, oral thrombin inhibitor. Recombinant
hirudin has been approved for limited clinical use.] Additional crosstalk
between the pathways of clotting is achieved by the FVIIA-TF-mediated
activation of the intrinsic pathway and the FXIIa-mediated activation of the
extrinsic pathway. The complete picture of physiologic blood clotting via the
formation of a hard fibrin clot is shown in Figure 34.15. The factors of the
clotting cascade are shown organized by function in Figure 34.16.
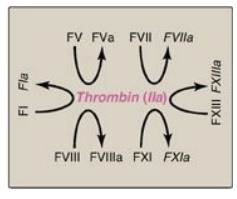
Figure 34.14 The importance of
thrombin in formation of the fibrin clot. a = active; F = factor.
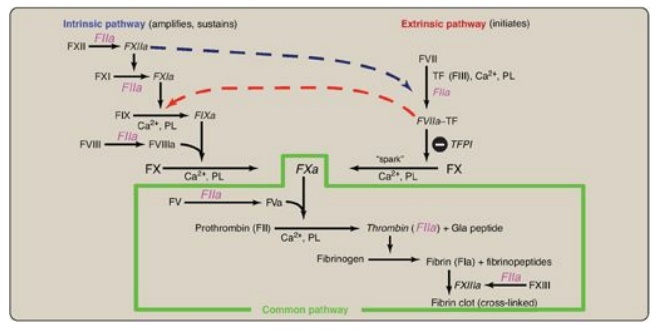
Figure 34.15 The complete
picture of physiologic blood clotting via the formation of a cross-linked
(hard) fibrin clot. a = active; F = factor; TF = tissue factor; TFPI = tissue
factor pathway inhibitor; PL = phospholipid; Gla = γ-carboxyglutamate.
Clinical laboratory tests are available to evaluate
the extrinsic through common pathways (prothrombin time [PT] using
thromboplastin and expressed as the International Normalized Ratio [INR]) and
the intrinsic through common pathways (activated partial thromboplastin time
[aPTT]). Thromboplastin is a combination of phospholipids + FIII. A derivative,
partial thromboplastin, contains just the phospholipid portion because FIII
isn’t needed to activate the intrinsic pathway.

Figure 34.16 Protein factors of
the clotting cascade organized by function. The activated form would be denoted
by an a after the numeral. [Note: Ca2+ is IV. There is no VI. I (fibrin)
is neither a protease nor an accesory protein. XIII is a transglutaminase.] Gla
= γ-carboxyglutamate.
Related Topics
