CIOMS II - Periodic Safety Updates
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: CIOMS Working Groups and their Contribution to Pharmacovigilance
This initiative was started in November 1989 at a time when several countries had requirements for periodic safety updates;
CIOMS 11 - PERIODIC SAFETY
UPDATES
RATIONALE
This
initiative was started in November 1989 at a time when several countries had requirements
for periodic safety updates; however, individual local regulatory authorities
were requesting that data (both foreign and domestic) be presented according to
different inclusion criteria, formats and time intervals. Due dates were often
determined by the national licensing approval date and therefore varied between
individual formulations of the same drug substance. Preparation of these
summarised safety updates had become a significant administrative burden for
manufacturers. Figure 23.1 shows the report preparation schedule for a
fictitious drug with different due dates and periods for review.
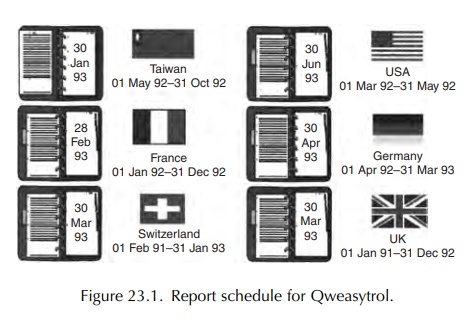
The
purpose of the CIOMS II working group was to explore the possibility of developing
a harmonized approach to preparing periodic safety updates that would meet most
existing needs and forestall any diversity in future requirements. It was also
hoped that if the guidelines on this approach were adequate and reasonable
other regulatory authorities would adopt them in the future. Standardisation
would also enable pharmacovigilance staff to focus on reviewing the data rather
than generating a battery of different reports.
PROCESS
The
working group undertook a survey of the currently existing requirements for
periodic safety updates, noting the diversity and identifying the ques-tions
which needed to be addressed in defining the content and format, and what might
be considered to be the essential elements. After considerable debate and compromise
on several controversial issues relat-ing to scope and content, a series of
proposals was then drafted in preparation for the pilot phase. Each
manufacturer representative undertook to draft a single prototype
summary-report on one of their own drugs using the proposed guidelines. Each
report was then sent personally to each regulator in the work-ing group and a
‘sanitised’ version was sent to the other manufacturer representatives. All
members of the working group took part in the critical evaluation of each pilot
report to examine the feasibility (data availability), resources required in
compilation and utility to the regulators of the information provided. On the
basis of the experiences gained in the pilot study, the guidelines were refined
and used to produce a model report on a fictitious drug (Qweasytrol) for
inclusion in the final report.
RECOMMENDATIONS
The
underlying principles of CIOMS II periodic safety updates were that they should
be prepared to stan-dard criteria that are practical and achievable, while
containing sufficient information to reassure regula-tors that the manufacturer
regularly reviewed its safety data. The safety updates should be as brief as
possible; it was recommended that the narrative content should not exceed about
10 pages. Data for all formulations of the same drug (including combination
products) should be included in one report and the same report should be
submitted at the same time to all regulatory authorities with a requirement for
safety updates.
Scope
The
proposal was that the guidelines should be applied to safety summaries produced
for all new chemical entities licensed for the first time in 1992. Subse-quent
updates would be based on 6-month interval data with cumulative data only
included where it gave a perspective on safety issues. Each subject drug would
have an international birth date (IBD), the first approval date for the first
formulation of the drug anywhere in the world, that would determine the date at
which 6-monthly reports commenced. A data-lock point (DLP) 6 months after the
IBD would be used to ‘freeze’ the database. Normally, the manufacturer should
make the report available within 45 calendar days of the DLP.
It
should be emphasised that periodic safety summaries were not intended for the
first communi-cation of urgent safety information. This should be reported
separately in the usual expedited manner.
Content
The
working group proposed that the periodic safety update was presented in nine
sections as follows:
• Introduction
• Core data sheet – the reference document for deter-mining ‘expectedness’
• The drug’s licensed status
• Update on regulatory or manufacturer actions taken for safety reasons
• Patient exposure
• Individual case histories (CIOMS line listing)
• Studies
o newly analysed studies containing important safety information
o targeted new safety studies
o published safety studies
• Overall safety evaluation
• Important information received after the DLP.
It was proposed that the individual case histories received during the 6-month period of review, and meeting specified criteria, should be presented in body system order of the most serious presenting sign or symptom in a CIOMS line-listing format. The criteria for case inclusion were as follows:
• unlabelled, serious attributable cases from studies (published or unpublished);
• all serious and non-serious unlabelled spontaneous reports (including relevant medically unconfirmed consumer reports);
• serious published case histories;
• serious cases from other sources (e.g. from regu-latory authorities).
The CIOMS line listing should consist of:
• Company reference number
• Country of origin of report
• Source of report (e.g. physician, literature)
• Age of patient
• Sex of patient
• Dose of drug
• Duration of treatment prior to event (time to onset)
• Description of reaction (as reported)
• Outcome.
A comment column was also suggested for use by the manufacturer to highlight important case infor-mation such as concurrent medication or underlying disease. It could also be used for the causality assess-ments (imputability) required by the French regulatory authority.
The overall safety evaluation should be a concise critical analysis and opinion explicitly including:
• increased frequency of known toxicity;
• drug interactions;
• overdose and its treatment;
• drug abuse;
• positive and negative experiences during preg-nancy and lactation;
• effects of long-term treatment;
• any specific safety issues relating to the treatment of special patient groups (e.g. elderly, children).
Finally, the evaluation should indicate whether the interim safety data remained in line with the cumu-lative experience to date or whether any modifica-tions were necessary to the company’s core safety information.
The CIOMS II report was published in 1992 (CIOMS, 1992).
INCORPORATION IN REGULATIONS
The
CIOMS II proposals for periodic safety updates were rapidly incorporated into
the European Draft Notice to Applicants but
with a few significant modifi-cations, including the concept of a European
rather than an international birth date. This effectively implied that periodic
safety reports currently scheduled to the IBD had to be rescheduled to the
first European approval date – a step away from the vision of harmonisation. A
European schedule for the frequency of submission was also included which
stated that 6-monthly reports were required for the first 2 years after
approval, followed by annual reports for 3 years and then 5-yearly there-after.
As individual countries began to implement their own periodic safety update
requirements they requested this schedule based on their own local approvals.
The scope of CIOMS II was also expanded to include all marketed products, not
just those approved in or after 1992.
Before
the European requirements could be finalised, ICH E2C adopted many of the CIOMS
II principles in the Clinical Safety Data
Manage-ment: Periodic Safety Update Reports for Marketed Drugs document
that reached Step 4 in November 1996
(ICH, 1996). This included further modifica-tions to the CIOMS II scope and
format, including some reordering of the sections and introduction of new
materials such as the summary tabulations to complement the line listing in
section 6. Figure 23.2 shows the ICH E2C table of contents and highlights the
changes from CIOMS II. There was also an addi-tional requirement to explain to
local regulators any differences between the local product information and the
company core safety information.

Fortunately,
ICH E2C reverted to the IBD for scheduling reports and the time for submission
after the DLP was increased to 60 days. However, while this may be achievable
for 6-monthly reports, there is concern because the ICH E2C format is now being
requested for periodic safety updates covering longer periods (including the
5-year reports for local product renewals in Europe).
While ICH E2C has been implemented in Japan and included in Volume IX of the Rules Governing Medicinal Products in the European Union – Notice to Marketing Authorisation Holders: Pharmacovigi-lance Guidelines, by 2005 the US Food and Drug Administration (FDA) had only issued draft peri-odic reporting requirements based on ICH E2C for consultation.
In
summary, the principles and guidelines proposed by CIOMS II achieved a
harmonised approach to preparing periodic safety updates that met most
exist-ing requirements in 1992. However, they were unable to forestall the
diversity of future requirements follow-ing their incorporation into regulatory
requirements around the world. In 2003 an addendum to ICH E2C was produced to
provide practical guidance for the preparation of periodic safety update
reports. This included many of the proposals on good summary reporting
practices recommended by the CIOMS V Working Group but the diversity of
requirements continues unabated.
Related Topics
