CIOMS III - Core Clinical Safety Information
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: CIOMS Working Groups and their Contribution to Pharmacovigilance
MS II introduced the concept of the core data sheet. It is a document prepared by the pharmaceutical manufacturer, containing the minimum essential safety information.
CIOMS III - CORE CLINICAL SAFETY
INFORMATION
RATIONALE
CIOMS
II introduced the concept of the core data sheet. It is a document prepared by
the pharmaceutical manufacturer, containing the minimum essential safety
information, such as ADRs, which the manufacturer stipulates should be listed
in all coun-tries where the drug is marketed (see Figure 23.3). It is also the
reference document by which ‘labelled’ and ‘unlabelled’ (or listedness and
unlistedness for ICH E2C) are determined. Thus, it should focus on the
important information required for rational clinical decision-making and
harmonise safety state-ments worldwide for public health and regulatory
purposes.
The
CIOMS III working group set out to propose principles and guidelines for
consistent decision-rules on the content of the Core Safety Information (CSI),
standard terms and definitions, and a standard format. One of the major
concerns was to minimise confusion among prescribers and other healthcare
professionals due to inconsistencies between the safety informa-tion presented
in different countries and by different manufacturers.
It
was therefore hoped that regulatory authori-ties would harmonise their basic
requirements for safety information in their local data sheets. However, the
working group acknowledged the possible need for cultural differences due to
medical and legal differences.
The
first edition of the CIOMS III report published in 1995 (CIOMS, 1995) focused
on CSI for marketed products, including the initial CSI that is prepared in
conjunction with the first market autho-risation submission, review and
approval. During CIOMS V discussions it was proposed that the same basic
philosophy and practices be applied to the safety information provided to
clinical investiga-tors during a development programme. The concept of
development core safety information (DCSI) as a discrete, focused section of
the Investigator’s Brochures, which would have the same format as, and would
evolve into, the CSI at initial marketing of the product, was therefore agreed.
A second edition of the CIOMS III report was issued in 1999 (CIOMS, 1999)
including the new proposals for Investigator’s Brochures.
PROCESS
The task of the working group was to develop propos-als for standard principles and guidelines addressing the what, when, how and where of CSI. The summary of product characteristics (SPC), the official document of the European Union, was used as a model to try to answer the following general questions:
• What evidence is needed, and how should it be used, to influence a decision on whether an adverse experience should be included, excluded or removed from the CSI?
• At what point in the accumulation and interpre-tation of information is the threshold crossed for inclusion or change in the CSI?
• What ‘good safety-labelling practices’ can be spec-ified concerning the clinical relevance of informa-tion, how it is expressed and the appropriateness of ‘class-labelling’?
• What should the sections of the CSI be called, how should they be defined and where should specific information be located?
At
the beginning of the process the group hoped to develop specific threshold
criteria, or an algorithm, for determining when information should be included
in the CSI. However, this was not possible and it became necessary to rely on
collective judgement to reach consensus. A series of case scenarios were
created from real-life examples for which the decision to amend a data sheet
was equivocal. Each member of the group was asked individually to make
decisions on the available data. In addition, each person was asked to list the
factors taken into consideration when reaching their conclusions. A total of 39
factors were identified and each member of the working group was asked to rank
the factors in order of importance. As expected, there was a considerable
divergence of opinion but overall the mostly highly ranked criterion for a
positive decision was the presence of positive rechallenge information. The
reader is referred to the original report for the remaining factors and their
respective rankings.
RECOMMENDATIONS
The
working group formulated a total of 65 propos-als relating to general
principles of good safety information and the what, when, how, where and who (responsibilities)
for CSIs. A selection of the most useful principles is given below.
What?
• The CSI should be determined by the needs of healthcare professionals in the context of a regula-tory and legal environment.
• Include what is practical and important to enable the prescriber to balance risks against benefit and to act accordingly.
• Avoid including events, especially minor events, that have no well-established relationship to therapy.
• There is a legal duty to warn but this must be balanced against the need to include only substan-tiated conclusions in the CSI.
• The CSI should include important information which physicians are not generally expected to know. (The converse is also true.)
When?
• As soon as relevant safety information becomes sufficiently well established it should be included in the CSI.
It
was not possible to define this more precisely but the working group introduced
the concept of ‘thresh-old’. This is dependent on the quality of information
available and the body and strength of the evidence according to the 39
criteria (plus two additional ones subsequently identified) in the ranking
exercise described above. Situations in which the threshold should be lowered
were identified. In general, infor-mation should be added sooner whenever it is
likely to help the physician make a differential diagnosis related to an
adverse event, spare extra tests, lead to the use of a specific targeted test
or facilitate early recognition of an event. Similarly, the thresh-old should
also be lowered if the ADR is medically serious or irreversible, if good
alternative drugs are available, a relatively trivial condition is being
treated, or the drug is being used for prophylaxis.
How?
• Keep ADRs identified in the initial CSI (pre-marketing experience) separate from those identi-fied subsequently.
• ADRs should be listed by frequency in body system order.
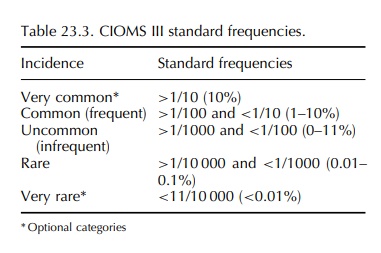
• Whenever possible, an estimate of frequency should be provided, expressed in a standard cate-gory of frequency.
While
the working group recognised that precise frequency rates can only be obtained
from studies and are limited to the more common reactions, it was agreed that
estimates of frequency in a standard format should be provided whenever
possible. Although it is difficult to estimate incidence on the basis of spon-taneous
reports due to the uncertainties in estimating denominator and the degree of
under-reporting, the group recommended the standard frequencies shown in Table
23.3.
Finally,
the working group defined the safety sections of the CSI, providing guidance on
the infor-mation which should be included in each section and outlined the
responsibilities of the company for remaining diligent and proactive, including
undertak-ing the scientific investigation of signals. The shared responsibility
of healthcare providers, patients, editors of medical journals and regulators
is also addressed.
INCORPORATION IN REGULATION
Since
the standards proposed by the CIOMS III working group would require continuous
evaluation, updating and refinement, it was suggested that they be retained as
guidelines and not adopted as regu-lations. They have been used as the basis of
the European Labelling Guidelines and many regulatory authorities have adopted
the standard categories of frequency. However, during the most recent
redraft-ing of the European Labelling Guidelines there was discussion regarding
their appropriateness when spon-taneous reports are the only source of data for
esti-mating frequency.
Related Topics
