Method
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: PEM in the UK
PEM is a non-interventional, observational cohort form of post-marketing surveillance.
METHOD
PEM
is a non-interventional, observational cohort form of post-marketing
surveillance. It is non-interventional because nothing happens to interfere
with the doctor’s decision regarding which drug to prescribe for each
individual patient. Thus, the method provides ‘real-world’ clinical data
involving neither inclusion nor exclusion criteria: the patients studied are
those who receive the drug in everyday medical practice. This ensures that the
data are generalisable.
In
the United Kingdom virtually all persons are registered with a general
practitioner (GP) who provides primary health care and issues prescriptions
(FP10s) for the medicines medically necessary. The patient takes the
prescription to a pharmacist who dispenses the medication and then sends the
FP10 to a central Prescription Pricing Division (PPD) which arranges the
pharmacist’s reimbursement. The Drug Safety Research Unit (DSRU) is, by virtue
of a long-standing and confidential arrangement, provided with electronic
copies of all those prescriptions issued nationally for the drugs being
monitored by PEM. These arrangements continue for a collection period which
allows exposure data to be collected for 20 000– 30 000 patients. For each of
these patients the DSRU prepares a computerised longitudinal record
compris-ing, in date order, all of the prescriptions for the monitored drug.
Thus, in PEM, the exposure data are national in scope throughout the collection
period and unaffected by the kind of selection and exclusion crite-ria that
characterise clinical trials. The exposure data are of drugs dispensed and
provided to the patient but there is no method of measuring compliance or the
use of non-prescription medication.
After
an interval of 3–12 (usually 6) months from the first prescription for each
individual patient the DSRU sends to the prescriber a ‘green form’
ques-tionnaire seeking information on any events that may have occurred since
the drug was first prescribed. An event is defined as any new diagnosis, any
reason for referral to a consultant or admission to hospital, any unexplained
deterioration (or improvement) in a concurrent illness, any suspected drug
reaction, any alteration of clinical importance in laboratory values, or any
other complaint which was considered of suffi-cient importance to enter in the
patient’s notes.
Information
which identifies the patient is deleted from the database when the green form
is received from the doctor. The doctor enters any number or code used in the
practice to identify the patient. This ensures that the clinical information
received by the DSRU is anonymised. The practice code or number is used if
follow-up information is sought from the doctor. In order to avoid placing an
unreasonable demand on GPs no more than four green forms are sent to each
doctor in any one month. The green form is illustrated in Figure 24.1, which
shows the other information requested of the doctor.

The
green form has been modified for certain studies with a small number of
additional questions (with yes, no, don’t know answers). These questions focus
on issues specific to the drug under study, for example the green form for the
PEM study on the NSAID meloxicam included questions about previ-ous history of
gastrointestinal conditions and intoler-ance to NSAIDs to identify possible
confounding by indication.
General
practitioners are not paid to fill in green forms. The arrangements allow good
contact between the doctor and the DSRU and this facilitates the collection of
any follow-up data that may be consid-ered necessary by the research physicians
monitoring each study and working within the DSRU. One of the strengths of PEM
is follow-up with the GP or the health service to obtain further information
from the doctor for a large number of reports. A list of reports for which
additional information is sought is included in Table 24.1.

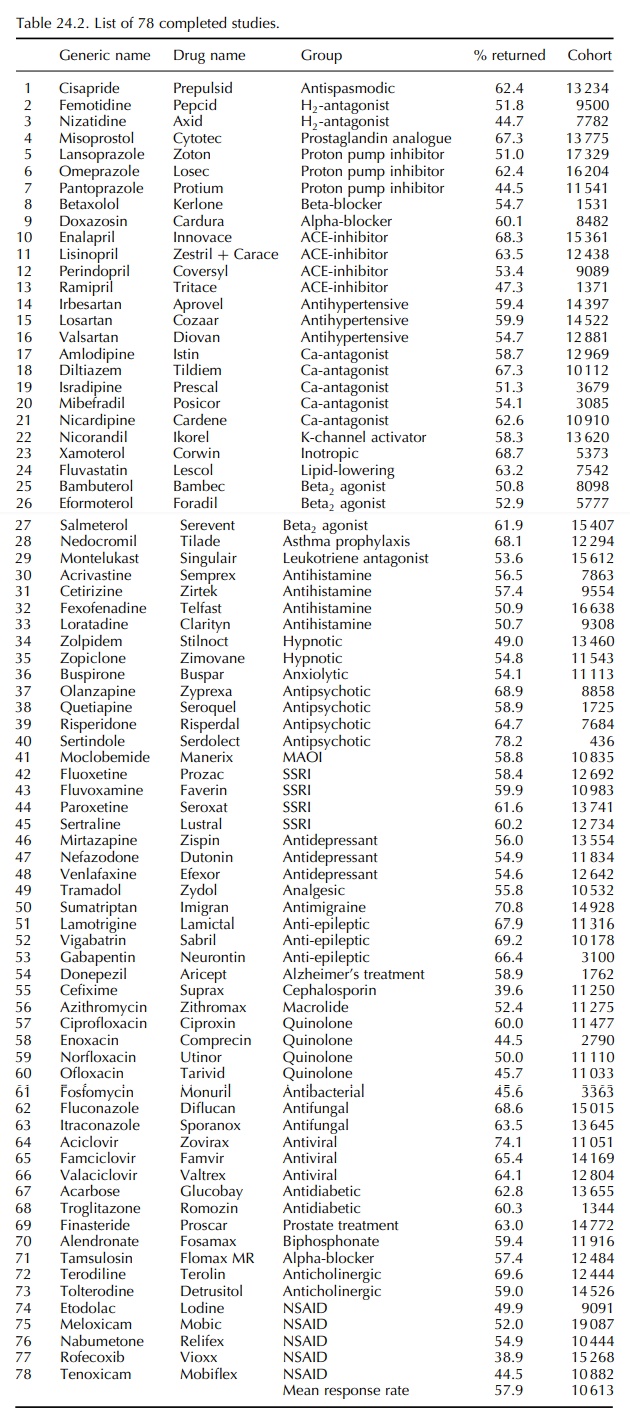
Over
the 78 studies listed in Table 24.2, an average of 58% of the green forms sent
out have been returned by the GPs to the DSRU. The cohort sizes, with an
average of 10 613 patients, as given in Table 24.2, are derived from the mean
52% of returned green forms which provide clinically useful data.
PEM
collects event data and does not ask the doctor to determine if any particular
event is due to an adverse drug reaction (ADR). If, however, the doctor does
consider the event to be an ADR or he has completed a yellow card (a
spontaneous ADR report) regarding the event, then he is asked to indicate this
on the green form.
Further
details of the methodology of PEM, includ-ing the methods of data coding,
computerisation and analysis, have been provided in a number of publica tions
(Inman, 1978b; Freemantle et al.,
1997; Mann et al., 1997).
Each
PEM study starts as soon as possible after the new drug has been marketed in
England. Each study aims to collect exposure and outcome data on approx-imately
10 000 patients. Some studies have included almost double that number and
attempts are now being made, when PEM is an ideal method for studying the early
experience with an important new drug, to maximise the size of the cohort. The
drugs included in the system are (as advocated by the Second Grahame-Smith
Working Party of the Committee on Safety of Medicines) those intended for
widespread, long-term use, special emphasis being given to drugs for which
treatment is likely to be both initiated and continued by the GP (Secretary of
State, 1986; BMA, 1996). In addition to drugs that are taken regularly, it has
also been possible to study products that are not used daily, such as
sildenafil for erectile dysfunction (Shakir et
al., 2001).
In
summary, the exposure data in PEM are derived from the prescriptions written by
GPs attending the individual patients; the outcome data are derived from the
green forms completed by those same GPs.
Within
the DSRU each green form questionnaire is scanned into the system and the image
is reviewed by a medical member of the DSRU staff so that impor-tant events can
be investigated. In addition to impor-tant events (Table 24.1), pregnancies and
deaths of uncertain cause are further investigated by the DSRU Research Fellows
who can, with the permission of the GP, access the patient’s life-time medical
records, death certificates, etc.
Interim
reports are written to summarise the data on each study with every 2500
patients entered into the database. These reports include a listing, by month
since the beginning of treatment, of all events reported. They are, if
possible, discussed with the Product Licence holder so that reporting
obligations to the regulatory bodies can be fulfilled. Wherever possible PEM is
undertaken in a collaborative but always independent relationship with the drug
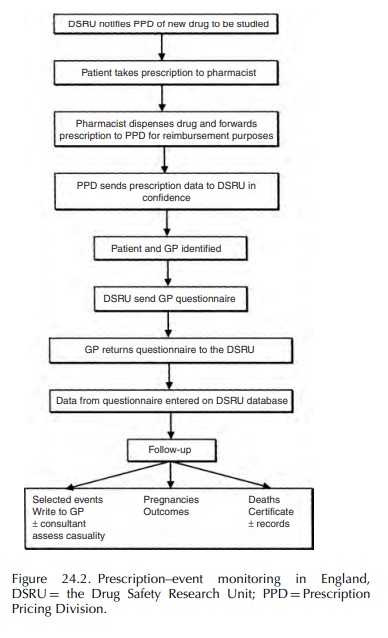
orig-inator. The methodology of PEM is summarised in Figure 24.2.
Related Topics
