Plasma protein binding
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacokinetics; Membrane Transport, Absorption And Distribution Of Drugs
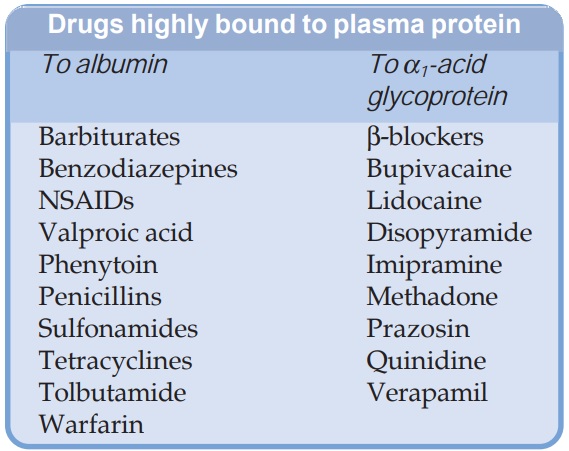
Most drugs possess physicochemical affinity for plasma proteins. Acidic drugs generally bind to plasma albumin and basic drugs to α1 acid glycoprotein. Binding to albumin is quantitatively more important.
PLASMA PROTEIN BINDING
Most
drugs possess physicochemical affinity for plasma proteins. Acidic drugs
generally bind to plasma albumin and basic drugs to α1 acid glycoprotein.
Binding to albumin is quantitatively more important. Extent of binding depends
on the individual compound; no generalization for a pharmacological or chemical
class can be made (even small chemical change can markedly alter protein binding),
for example the binding percentage of some benzodiazepines is:
Flurazepam 10% Alprazolam 70%
Lorazepam 90% Diazepam 99%
Increasing
concentrations of the drug can progressively saturate the binding sites:
fractional binding may be lower when large amounts of the drug are given. The
generally expressed percentage binding refers to the usual therapeutic plasma
concentrations of a drug. The clinically significant implications of plasma
protein binding are:
Highly plasma protein bound drugs are largely restricted to the
vascular compartment because protein bound drug does not cross membranes
(except through large paracellular spaces, such as in capillaries). They tend
to have smaller volumes of distribution.
The bound fraction is not available for action. However, it is
in equilibrium with the free drug in plasma and dissociates when the
concentration of the latter is reduced due to elimination. Plasma protein
binding thus tantamounts to temporary storage of the drug.
High degree of protein binding generally makes
the drug long acting, because bound fraction is not available for metabolism or
excretion, unless it is actively extracted by liver or kidney tubules.
Glomerular filtration does not reduce the concentration of the free form in the
efferent vessels because water is also filtered. Active tubular secretion,
however, removes the drug without the attendant solvent → concentration of free
drug falls → bound drug
dissociates and is eliminated resulting in a higher renal clearance value of
the drug than the total renal blood flow (See
Fig. 3.2). The same is true of active transport of highly extracted drugs in
liver. Plasma protein binding in this situation acts as a carrier mechanism and
hastens drug elimination, e.g. excretion of penicillin; metabolism of
lidocaine. Highly protein bound drugs are not removed by haemodialysis and need
special techniques for treatment of poisoning. Generally expressed plasma
concentrations of the drug refer to bound as well as free drug. Degree of
protein binding should be taken into account while relating these to concentrations
of the drug that are active in vitro,
e.g. MIC of an antimicrobial.
One drug can bind to
many sites on the albumin molecule. Conversely, more than one drug can bind to
the same site. This can give rise to displacement interactions among drugs bound
to the same site(s): drug bound with higher affinity will displace that bound
with lower affinity. If just 1% of a drug that is 99% bound is displaced, the
concentration of free form will be doubled. This, however, is often transient
because the displaced drug will diffuse into the tissues as well as get
metabolized or excreted: the new steady state free drug concentration is only
marginally higher unless the displacement extends to tissue binding or there is
concurrent inhibition of metabolism and/or excretion. The overall impact of
many displacement interactions is minimal; clinical significance being attained
only in case of highly bound drugs with limited volume of distribution (many
acidic drugs bound to albumin) and where interaction is more complex. Moreover,
two highly bound drugs do not necessarily displace each other—their binding
sites may not overlap, e.g. probenecid and indomethacin are highly bound to albumin
but do not dis place each other. Similarly, acidic drugs do not generally
displace basic drugs and vice versa.
Some clinically important displacement interactions are:
Salicylates displace sulfonylureas.
Indomethacin, phenytoin displace warfarin.
Sulfonamides and vit K displace bilirubin
(kernicterus in neonates).
Salicylates displace methotrexate.
In hypoalbuminemia, binding may be reduced and
high concentrations of free drug may be attained, e.g. phenytoin and
furosemide. Other diseases may also alter drug binding, e.g. digitoxin,
phenytoin and pethidine binding is reduced in uraemia; propranolol binding is
increased in pregnant women and in patients with inflammatory disease (acute
phase reactant α1 acidglycoprotein
increases).
Skeletal muscle, heart — digoxin, emetine (bound to muscle proteins).
Liver — chloroquine, tetracyclines, emetine, digoxin.
Kidney — digoxin, chloroquine, emetine.
Thyroid — iodine.
Brain — chlorpromazine, acetazolamide, isoniazid.
Retina — chloroquine (bound to nucleoproteins).
Iris — ephedrine, atropine (bound to melanin).
Bone and teeth — tetracyclines, heavy metals (bound to
mucopolysaccharides of connective tissue), bisphosphonates (bound to hydroxyapatite)
Adipose tissue — thiopentone, ether, minocycline, phenoxybenzamine, DDT dissolve
in neutral fat due to high lipidsolubility; remain stored due to poor blood
supply of fat.
