Distribution
| Home | | Pharmacology |Chapter: Essential pharmacology : Pharmacokinetics; Membrane Transport, Absorption And Distribution Of Drugs
Once a drug has gained access to the blood stream, it gets distributed to other tissues that initially had no drug, concentration gradient being in the direction of plasma to tissues.
DISTRIBUTION
Once a drug has gained
access to the blood stream, it gets distributed to other tissues that initially
had no drug, concentration gradient being in the direction of plasma to
tissues. The extent of distribution of a drug depends on its lipid solubility,
ionization at physiological pH (a function of its pKa), extent of binding to
plasma and tissue proteins, presence of tissuespecific transporters and
differences in regional blood flow. Movement of drug proceeds until an equilibrium
is established between unbound drug
in plasma and tissue fluids.
Subsequently, there is a parallel decline in both due to elimination.
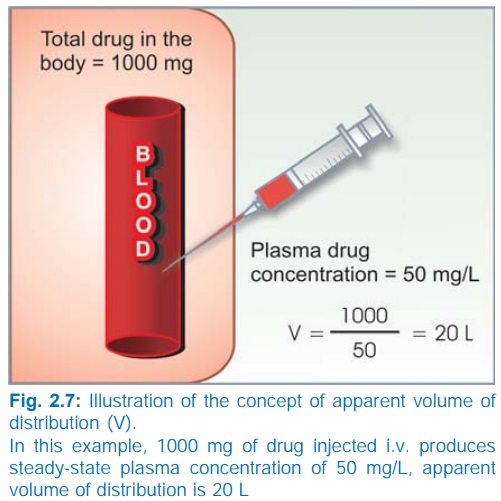
Apparent volume of distribution (V) Presuming that the body behaves as a single
homogeneous compartment with volume V
into which drug gets immediately and uniformly distributed
dose administered i.v.
V = ——————————– ...(3)
plasma concentration
Since in the example
shown in Fig. 2.7, the drug does not actually distribute into 20 L of body
water, with the exclusion of the rest of it, this is only an apparent volume of
distribution which can be defined as “the volume that would accommodate all the
drug in the body, if the concentration throughout was the same as in plasma”.
Thus, it describes the amount of drug present in the body as a multiple of that
contained in a unit volume of plasma. Considered together with drug clearance,
this is a very useful pharmacokinetic concept.
Lipidinsoluble drugs
do not enter cells— V approximates
extracellular fluid volume, e.g. streptomycin,
gentamicin 0.25 L/kg.
Distribution is not
only a matter of dilution, but also binding and sequestration. Drugs extensively
bound to plasma proteins are largely restricted to the vascular compartment and
have low values, e.g. diclofenac and warfarin (99% bound) V = 0.15 L/kg.
Drugs sequestrated in
other tissues may have, V much more
than total body water or even body mass,
e.g. digoxin 6 L/kg, propranolol 4 L/kg, morphine 3.5 L/kg, because most of the
drug is present in other tissues, and plasma concentration is low. Therefore,
in case of poisoning, drugs with large volumes of distribution are not easily
removed by haemodialysis.
Pathological states,
e.g. congestive heart failure, uraemia, cirrhosis of liver, etc. can alter the V of many drugs by altering distribution
of body water, permeability of membranes, binding proteins or by accumulation
of metabolites that displace the drug from binding sites.
More
precise multiple compartment models for drug distribution have been worked out,
but the single compartment model, described above, is simple and fairly
accurate for many drugs.
More precise multiple compartment models for drug distribution
have been worked out, but the single compartment model, described above, is
simple and fairly accurate for many drugs.
Factors Governing Volume of Drug Distribution
Lipid: water partition
coefficient of the drug
pKa value of the drug
Degree of plasma
protein binding
Affinity for different
tissues
Fat: lean body mass
ratio, which can vary with age, sex, obesity, etc.
Diseases
like CHF, uremia, cirrhosis
Redistribution
Highly lipidsoluble drugs get initially distributed to organs with high
blood flow, i.e. brain, heart, kidney, etc. Later less vascular but more bulky
tissues (muscle, fat) take up the drug—plasma concentration falls and the drug
is withdrawn from these sites. If the site of action of the drug was in one of
the highly perfused organs, redistribution results in termination of drug
action. Greater the lipid solubility of the drug, faster is its redistribution.
Anaesthetic action of thiopentone sod. injected i.v. is terminated in few
minutes due to redistribution. A relatively short hypnotic action lasting 6–8
hours is exerted by oral diazepam or nitrazepam due to redistribution despite
their elimination t ½ of > 30 hr. However, when the same drug is given
repeatedly or continuously over long periods, the low perfusion high capacity
sites get progressively filled up and the drug becomes longer acting.
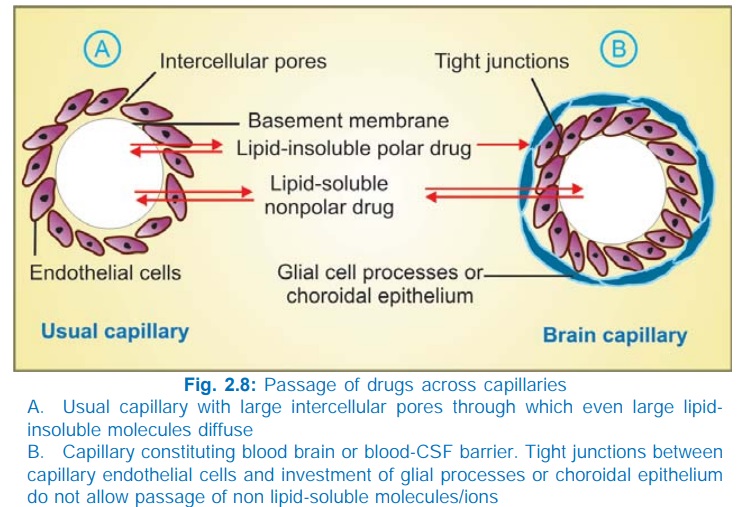
Penetration into brain and CSF
The capillary endothelial cells in brain have tight
junctions and lack large intercellular pores. Further, an investment of neural
tissue (Fig. 2.8B) covers the capillaries. Together they constitute the so
called bloodbrain barrier. A similar bloodCSF barrier is located in the choroid plexus: capillaries are lined by choroidal
epithelium having tight junctions. Both these barriers are lipoidal and limit
the entry of nonlipidsoluble drugs, e.g. streptomycin, neostigmine, etc. Only
lipidsoluble drugs, therefore, are able to penetrate and have action on the
central nervous system. In addition, efflux transporters like Pgp and anion
transporter (OATP) present in brain and choroidal vessels extrude many drugs
that enter brain by other processes. Dopamine does not enter brain but its precursor
levodopa does; as such, the latter is used in parkinsonism. Inflammation of meninges
or brain increases permeability of these barriers. It has been proposed that
some drugs accumulate in the brain by utilizing the transporters for endogenous
substances.
There
is also an enzymatic bloodbrain barrier: monoamine oxidase (MAO), cholinesterase
and some other enzymes are present in the capillary walls or in the cells
lining them. They do not allow catecholamines, 5HT, acetylcholine, etc. to
enter brain in the active form.
The
bloodbrain barrier is deficient at the CTZ in the medulla oblongata (even lipidinsoluble
drugs are emetic) and at certain periventricular sites—(anterior hypothalamus).
Exit of drugs from the CSF and brain, however, is not dependent on lipidsolubility
and is rather unrestricted. Bulk flow of CSF (alongwith the drug dissolved in
it) occurs through the arachnoid villi and nonspecific organic anion and cation
transport processes (similar to those in renal tubule) operate at the choroid
plexus.
Passage across placenta
Placental membranes
are lipoidal and allow free passage of lipophilic drugs, while restricting
hydrophilic drugs. The placental efflux Pgp also serves to limit foetal
exposure to maternally administered drugs. However, restricted amounts of
nonlipidsoluble drugs, when present in high concentration or for long periods
in maternal circulation, gain access to the foetus. Some influx transporters also
operate at the placenta. Thus, it is an incomplete barrier and almost any drug
taken by the mother can affect the foetus or the newborn (drug taken just
before delivery, e.g. morphine).
