Post-Marketing Experience 1977–82
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Nomifensine and Haemolytic Anaemia
Unit sales are shown in terms of defined daily doses of 100 mg.
POST-MARKETING EXPERIENCE
1977–82
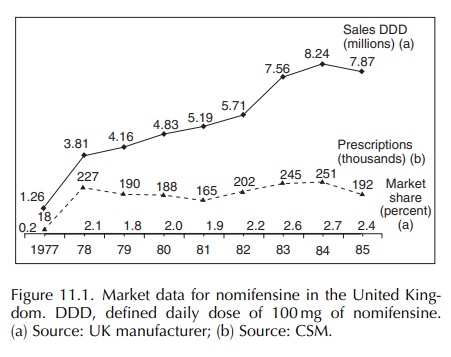
Figure
11.1 shows the market data for nomifensine in the United Kingdom. Unit sales
are shown in terms of defined daily doses of 100 mg. This terminology was not
routinely used in 1979–80 and was only adopted by the World Health Organisation
(WHO) in January 1992 as an international standard denominator for calculating
incidence. The numbers of prescrip-tion were provided by the Committee on
Safety of Medicines (CSM) from the Prescription Pric-ing Authority, and the
percentage UK market share achieved by nomifensine is shown; the total
repre-sents all antidepressant prescribing including generic compounds.
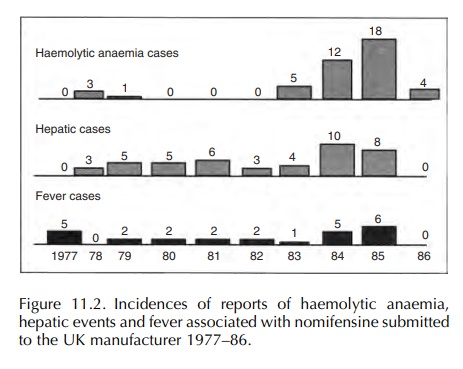
Figure
11.2 shows the incidence of reports of haemolytic anaemia, hepatic events and
fever over time.
Nomifensine
was first marketed as a 25 mg capsule formulation on 10 October 1977, whereas
the 50 mg capsule was made available on 1 January 1979. Between 1978 and 1979,
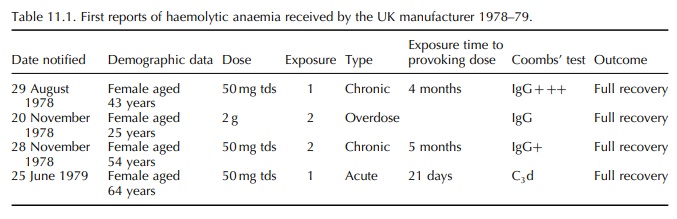
four reports of acute or chronic haemolytic anaemia occurring during treat-ment
with nomifensine were received by the manu-facturer (Table 11.1). The patients
were females with an age range of 25–64 years. Three of them were taking 150 mg
nomifensine daily. Each had a different history of exposure and onset of
haemolytic anaemia.
The
type of haemolytic anaemia was characterised as chronic or acute, depending on
the pattern of symp-toms, their severity and the presence or absence of
intravascular haemolysis. The symptoms of chronic-onset haemolytic anaemia
included lethargy, fatigue and breathlessness, whereas the acute presentation
of the condition involved backache, loin pain, jaundice and haematuria and, in
certain cases, fever, renal failure and cardiorespiratory collapse. The Coombs’
(anti-globulin) test was positive in all four cases. All of the patients had
received concomitant medication, although it was considered to be
non-contributory. When nomifensine was stopped, the patients made a full and
uneventful recovery.
The
first documented report of haemolytic anaemia, published in the Lancet, came from France. This was a
case of immune haemolytic anaemia and acute renal failure in a 50-year-old
woman, who was diagnosed in May 1978 (Bournerias and Habibi, 1979). She had had
seven episodes of malaise, chills, pain and fever of 2–4 h duration that were
accompanied by dark urine and transient jaundice.
During
one of the episodes in July 1978, she had had oliguria. At this time, she had a
positive Coombs’ test and a haemoglobin level of 10 g/dl. Before the episode,
she had been treated for an unrelated illness with levomepromazine, diazepam
and nomifensine. She made an uneventful recovery on stopping the medication.
The serum of the patient demonstrated an antibody that agglutinated red blood
cells only in the presence of nomifensine. The authors called for immunological
studies for anti-nomifensine antibod-ies in patients on long-term treatment.
Another
case of acute haemolysis and renal failure (following an overdose of
nomifensine) was published the following year (Prescott et al., 1980) (Table 11.1), and three others from outside the
United Kingdom were published in 1981–82 (Eckstein et al., 1981; Habibi et al.,
1981). One of these cases had intravas-cular haemolysis during treatment with
nomifensine (Lyllof et al., 1982).
Although
these reports were of concern, it was not considered at the time that
nomifensine was more liable to cause haemolytic anaemia than other marketed
drugs. However, heightened vigilance was recommended, and the manufacturer
initiated many retrospective and prospective immunological stud-ies. These
investigations failed to provide support for a cause-and-effect relationship
between nomifen-sine and haemolytic anaemia. Some patients with haemolytic
anaemia had a negative Coombs’ test, whereas other patients with a positive
Coombs’ test did not have haemolysis. Nevertheless, in view of the suspected
link between the antidepressant and the blood dyscrasias, haemolytic anaemia
was included among the side effects listed in the January 1981 data sheet.
Between
1981 and 1982, there were three more UK cases of haemolytic anaemia reported to
the Depart-ment of Health and Social Security (DHSS). They had not been
referred to the manufacturer. They occurred among patients who had received a
total of 990 000 prescriptions for nomifensine. This suggested an inci-dence of
only about 1 per 150 000 patients, and thus no regulatory action was considered
necessary (CSM Update, 1986; Mann, 1988).
These reports did not provide a consistent basis for any general announcement concerning the safety of nomifensine from the company or from a regulatory authority. They placed nomifensine at worst with a group of marketed drugs associated with haemolytic anaemia. This included stibophen, quinidine, paracetamol, penicillin, sulphonamides, tolbutamide, chlorpromazine, tetracycline, cephalosporins, insulin, rifampicin, hydralazine, streptomycin, triamterene and probenecid for immune haemolytic anaemia, and amongst methyldopa, mefenamic acid, flufe-namic acid and levodopa for autoimmune haemolytic anaemia.
Nevertheless,
the company acted on the reports to institute both retrospective and
prospective studies in Germany, France, the United Kingdom and Austria, to
determine potential groups at risk. Between January 1979 and June 1980, 312
patients in these studies who had been treated for more than 3 months with
nomifensine were given a Coombs’ test, and sera from 220 patients were
subjected to intensive immuno-logical investigations. Even with these studies,
the results did not prove a causative link with nomifen-sine. The Coombs’ test
proved to be inappropriate as a prediction of possible groups at risk amongst
nomifen-sine users. Some patients without haemolysis had a positive Coombs’
test, and later several patients with haemolytic anaemia were found to have a
negative Coombs’ test.
In
the course of time, supportable evidence for attributing haemolytic anaemia to
nomifensine was produced, and in January 1981, this addition to the UK data
sheet was agreed: ‘Haemolytic anaemia has also been reported in rare cases as
has a rise in body temperature’. This also appeared in the ABPI Data Sheet Compendium in
October 1981.
Concern
over the occurrence of haemolytic anaemia and the other serious reactions led
to many additional immunological investigations, and this work in due course provided
further evidence for the immunolog-ical basis of the haemolytic anaemia
reaction (Walti et al., 1983;
Miescher, 1985; Salama and Mueller-Eckhardt, 1985).
Salama
et al. (1984) demonstrated a
nomifensine-dependent antibody that reacted exclusively to its ex vivo antigen (fresh serum of a
volunteer who had taken a therapeutic
dose of the drug) but not to nomifensine itself. The investigators later showed
an ‘extraordinary heterogeneity’ of antibody response following the ingestion
of the antidepressant. Of 19 samples, only 5 were primarily reactive to
nomifen-sine. The majority reacted in the presence of one or more metabolites
and ex vivo antigens, indi-cating
specificity for an unidentified early or late metabolite.
All
samples belonged to the immunoglobulin G (IgG) or IgM class or both and were
capable of activating complement. At least one sample had two
nomifensine-dependent red blood cell antibodies, whereas one had platelet
antibodies. The latter explained the occurrence of purpura alongside the haemolysis.
It is of interest that 7 of the 19 patients had also signs of transient renal
insufficiency, whereas 6 had increased levels of serum transaminase (type not
specified; Salama and Mueller-Eckhardt, 1985).
Previously
(in September 1978, published April 1979), the data sheet had been amended to
draw atten-tion to the association of nomifensine with fever. There had been
several reports of this in Germany, and five reports were submitted to the UK
manufac-turer in 1977. The data sheet stated that there had been ‘rare cases of
rise in body temperature which returned to normal when the drug was withdrawn’.
The
data sheet of 1981 also drew attention to the association of nomifensine with
changes in liver enzymes by stating that :
In rare cases,
increases in liver enzymes (serum transaminases and alkaline phosphatase) have
been observed.
Because
of receiving four reports of haemolytic anaemia in 1978–79, the manufacturer
undertook the following actions:
·
Full investigation of each case report. The normal company’s
operating procedure involved acquir-ing full information on each case from the
prescribing doctor, if necessary visiting the doctor to discuss the case and
being accompanied on such visits by medical personnel from the central drug
safety department of the company headquarters.
·
All cases to be reported to the parent company and the UK
DHSS.
·
Re-appraisal of all preclinical work and clinical trials to
see whether there was any evidence of blood dyscrasias. None was found.
·
Retrospective and prospective immunological studies. These
produced no consistent results related to the clinical use of the drug.
·
Sales representatives to be informed of publica-tions and
investigative activities to respond appro-priately to enquiries.
·
Data sheet changes with international agreement relating to
fever, haemolytic anaemia and the liver.
Related Topics
