Post-Marketing Experience 1983–86
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Nomifensine and Haemolytic Anaemia
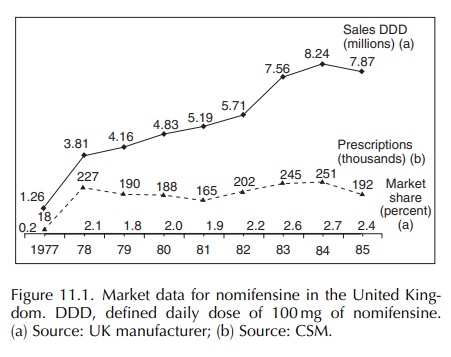
The increasing incidence of haemolytic anaemia from 1983 might appear to have been related to the launch of the 100 mg single daily dose formulation on 31 January 1983.
POST-MARKETING EXPERIENCE
1983–86
The
increasing incidence of haemolytic anaemia from 1983 might appear to have been
related to the launch of the 100 mg single daily dose formulation on 31 January
1983 (Figure 11.1). However, no evidence emerged to support this. It appears
that new additional sales were generated by this launch and that the
asso-ciated promotion may have made doctors more aware of nomifensine.
Prescriptions, sales and market share increased in 1983 by 21%, 32% and 18%,
respec-tively. This, together with the data sheet changes and literature
reports, may have served to alert doctors to the association of unusual
symptoms with the use of nomifensine.
Reports
of other severe untoward events that could have had an immunological basis also
appeared in the literature in 1984–85: thrombocytopenia (Green et al., 1984), hepatitis (Vaz et al., 1984), alveolitis (Hamm et al., 1985) and a systemic lupus
erythematosus (SLE)-like reaction
(Garcia-Morteo and Maldonado-Cocco, 1983; Schönhöfer and Groticke, 1985). Those
appearing in the British medical literature could possibly have contributed to
an increased awareness amongst prescribers of adverse events associated with the
drug. The first fatal case of immune haemolysis was published in 1985 (Sokol et al., 1985), and two other cases were
reported later the same year (Hamm et al.,
1985; Schönhöfer and Groticke, 1985).
In
the early to mid-1980s following the withdrawal of benoxaprofen and the
recognition of problems with other non-steroidal anti-inflammatory drugs,
together with promotion of the government’s Yellow Card scheme, there was an
increasing acceptance amongst doctors of the need to report adverse experiences
with commonly prescribed drugs. In September 1983, the antidepressant zimeldine
was withdrawn from the market following the identification of a serious
neurological disorder, the Guillain–Barré syndrome. The publicity given to this
may have affected the reporting of adverse events to drug therapy, including
nomifensine.
The
purpose of showing the comparative incidences of fever, hepatic reactions and
haemolytic anaemia in Figure 11.2 is not to suggest any common underly-ing
pathology to these three conditions; none has ever been substantiated. It is to
indicate that, whilst report-ing rates of haemolytic anaemia and hepatic
problems (enzyme changes, jaundice or hepatitis) significantly increased with
time, this was not the case with reports of febrile reactions. The incidence of
these never reached the same levels as in some other countries, e.g. Germany.
Cases in the United Kingdom in which fever was associated with haemolysis were
catalogued in the haemolytic anaemia group.
Table
11.2 shows the UK manufacturer’s total database of 296 events; this is to be
compared with the CSM’s Yellow Card database of 543 suspected adverse
reactions. The company had 45 reports of haemolytic anaemia of which 43 were
thought to be associated with the drug. This is to be compared with the CSM’s
59 reports of which 49 contained suffi-cient information to attribute
nomifensine as the prob-able, or a possible, cause. Forty-five of the 49 (92%)
patients were women, although females received only 71% of the prescriptions
for the drug. Some of the subjects, who had had a previous course of
nomifensine without experiencing unwanted effects, developed acute haemolytic
anaemia on recommenc-ing treatment, whereas others developed haemolytic anaemia
after months or years of continuous use. In 18 patients, the haemolysis was
severe: 11 of them devel-oped renal failure and 4 died. Although haemolytic
anaemia was the most frequently reported serious adverse reaction, concern was
also expressed over other untoward effects (CSM Update, 1986).

From
1983 onwards, there was a steady rise in the number of reports of haemolytic
anaemia to the UK manufacturer, with 5 reports in 1983, 12 in 1984 and 18,
including 3 fatalities, in 1985. The first nomifensine-associated fatality in
the United King-dom was reported on 10 February, the second was reported on 31
March and the third on 10 April 1985. The three cases were discussed with the
DHSS on 1 May 1985.
The
first of these fatal cases was published in the British Medical Journal in August 1985 (Sokol et al., 1985). The patient was a 36-year-old female who collapsed 1 h after taking one 100
mg tablet. She had been treated with nomifensine for 1 week but stopped taking
it because of dizziness. There was no jaundice or haematuria. On examination, she
was conscious but pale, cyanosed and shocked. Her blood pressure was 90/50
mmHg, and her pulse was 90/min. Haematological tests showed spontaneous red
cell agglutination, with free haemoglobin in the plasma, and the following
results: haemoglobin 5 g/dl, biliru-bin 4 mol/l and lactate dehydrogenase 1071
IU/l. The patient had severe acidosis. Acute intravascular haemolysis was
diagnosed. Attempts at resuscitation failed, and the patient died.
Immunological investiga-tions showed a positive Coombs’ test with antisera to
IgG, IgM and Cl. The serum contained cold-reacting auto-antibodies and pan
antibodies. In the presence of nomifensine, the antibodies led to the
agglutination of red cells.
The proposed mechanism was that drug and anti-body combined to form loose immune complexes that attached themselves to the red cells and activated complement. Complement activation led to haemol-ysis, disseminated intravascular coagulation and the shock-lung syndrome.
Between
January 1983 and mid-June 1985, the DHSS was aware of 29 reports of haemoly-sis
in 592,000 prescriptions—approximately one in 1:20,000 prescriptions (CSM
Update, 1986).
In
July 1985, the CSM’s bulletin, Current
Prob-lems, highlighted the dangers of newer antidepressants and presented a summary of adverse drug
reactions to nomifensine. A new data sheet was published with information
submitted in October 1984. This stated that :
In rare cases,
haemolytic anaemia and abnormal liver function tests with or without clinical
jaundice have been observed. These reactions subside within a short time of
discontinuing Merital (nomifensine) but may recur if it is taken again.
In
September 1985, there were joint discussions between the company and the DHSS
on a complete revision of the data sheet. On 24 September, the current data
sheet was put in abeyance pending the outcome of these discussions and all
promotion of nomifensine ceased.
On
30 September 1985, the company issued a ‘Dear Doctor’ letter warning of the
serious adverse reactions reported internationally; this letter was a version
of a similar ‘Red Hand’ letter issued at the same time by the parent company in
Germany.
On
7 December 1985, the ‘CSM Update’ on antide-pressants, published in the British Medical Journal, summarised the
comparative adverse reaction reports on all antidepressants (CSM Update, 1985).
On
16 December 1985, the Drug and
Thera-peutics Bulletin published an article ‘Trouble with nomifensine’ after several revisions
since the first draft in May. This was followed by many newspaper reports on
the drug.
Between
mid-June and the end of November 1985, the DHSS was aware of 25 reports of
haemolysis in 96,000 prescriptions (1:4000; CSM Update, 1986). This was the
first time that the incidence had increased to a level above 1:10,000 (the
accepted WHO defi-nition of a rare incidence), giving rise to a situation in
which the benefits of the drug could no longer be said to outweigh the risks of
haemolytic anaemia.
Four
further cases of haemolytic anaemia were reported to the company in January
1986. One of these patients subsequently died. The UK data contributed to the
ongoing appraisal of nomifensine being under-taken by the parent company, and
this led to the product’s withdrawal from worldwide markets on 22 January 1986.
Table
11.3 summarises the events and assessments leading to the withdrawal of
nomifensine 10 years after its first market launch.

Related Topics
