Use of IR Spectroscopy for Structure Determination
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Structure Determination of Organic Compounds
As seen above, IR spectroscopy is most commonly used to identify functional groups and bonding patterns in molecules from the higher energy portion of the spectrum where absorptions are primarily due to bond-stretching vibrations.
USE OF IR SPECTROSCOPY FOR
STRUCTURE DETERMINATION
As
seen above, IR spectroscopy is most commonly used to identify functional groups
and bonding patterns in molecules from the higher energy portion of the
spectrum (1200 – 4000 cm−1) where absorptions are primarily due to bond-stretching
vibrations. Some information on atom connectivity in the molecule can also be
deduced from the frequency shifts caused by structural factors. In general,
however, it is not possible to completely deduce the structure of a molecule by
examination of its IR spectrum. However, IR spectroscopy is a powerful
complement to NMR spectroscopy for structure determination.
For
example, the reaction of 3-chlorocyclohexanone with DBU in toluene gives a
product which is seen to have two vinyl protons by NMR and thus is an
elimination product, probably either A or B. Now while one might rationalize by
both chemical intuition and by the splitting pattern that conjugated isomer A
is the product, examination of the IR spectrum shows a carbonyl group (at 1680
cm−1) and an olefin band
(at 1630 cm−1). A typical
cyclohexanone comes at 1710 cm−1 and cyclohexene
comes at 1643 cm−1.
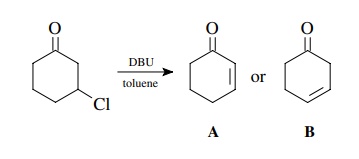
Clearly
the observed frequencies of the product are at lower frequencies than a simple
ketone or olefin and are indicative of a conjugative interaction between these
two functions. Thus A and not B is the product.
Treatment
of propiophenone with m-CPBA (m-chloroperbenzoic acid) in
dichloromethane gives a single product. The carbonyl absorption of
pro-piophenone is at 1695 cm−1 whereas the product
has a carbonyl absorption at 1745 cm−1. This information
reveals that the carbonyl group is intact but is no longer a ketone. The shift
to higher frequency is consistent with the conversion to an ester, and thus the
product could be either C or D.
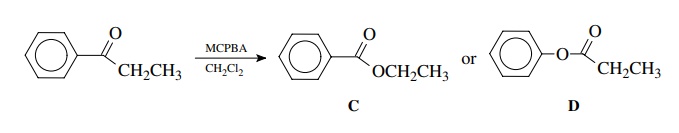
Since
ethyl benzoate C has a carbonyl
stretch at 1725 cm−1, the likely product is D,
phenyl propionate. This is confirmed by the NMR spectrum, which has the
methylene group as a quartet at 2.42δ.
This chemical shift is typical for a methylene group next to an ester carbonyl
but is much too high field for the –CH2–group in ethoxy ester C, which comes at about 3.6δ.
Reaction
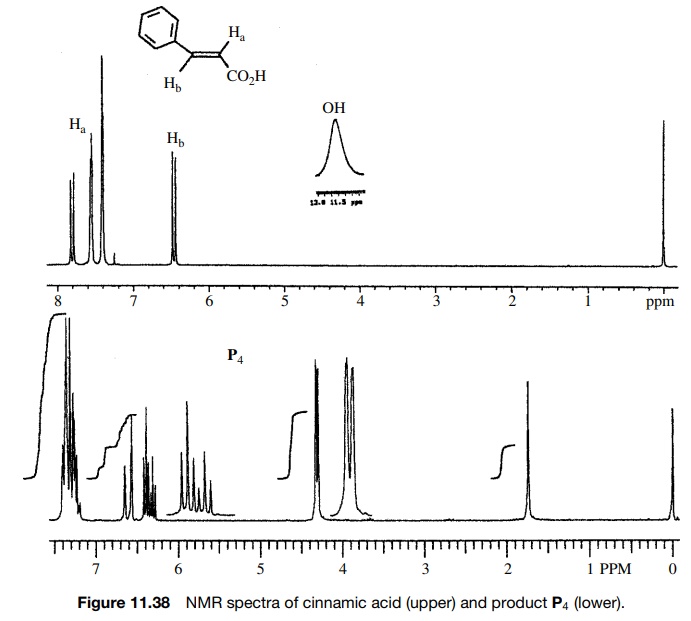
of cinnamic acid, which has the IR and 1H NMR spectra shown in
Figures 11.36 – 11.38, with BH3·THF gives rapid consumption of the
starting material. A single product P4
is formed that has the spectra shown. Comparison of the IR spectra shows that
the double bond in the reactant (1630 cm−1) is intact in the
product (1654 cm−
1) but moved to higher frequency. The product P4 also has both vinylic (3026 cm−1) and saturated
(2861 cm−1) C–H bonds. Both reactant and product have an O–H
adsorption, but the strong H bonding in
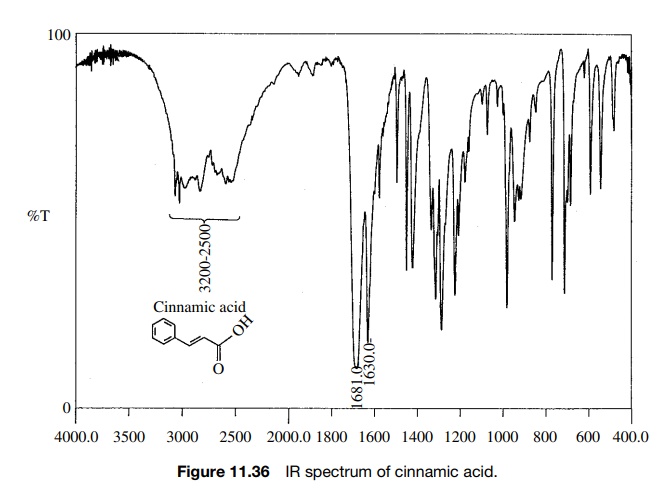
Furthermore the carbonyl group in the
reactant acid (1681 cm−1) is missing in the product. The IR data suggest that the
carboxylic acid has been reduced by BH3·THF in preference to hydroboration
of the double bond.
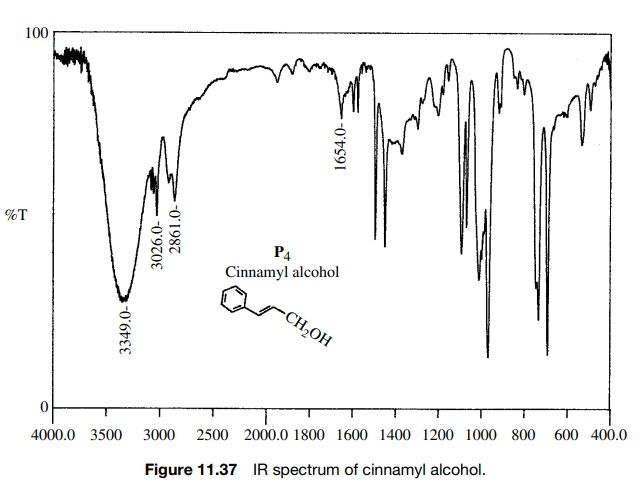
The
1H NMR corroborates this conclusion since two vinyl protons are
observed both in the reactant and product; however, a new two-proton doublet
appears at 4.15δ for the newly produced
allylic methylene group. The acid O–H pro-ton is moved far upfield as well. The
coupling constants of the vinyl protons (J
= 16 Hz) show the
starting compound to be trans, and the large splitting for the downfield vinyl
doublet of the product (J = 16 Hz) shows the trans
stereochemistry to be maintained in the unsaturated alcohol product. Moreover
the splitting between the methylene group and the upfield vinyl proton clearly
supports its allylic position.
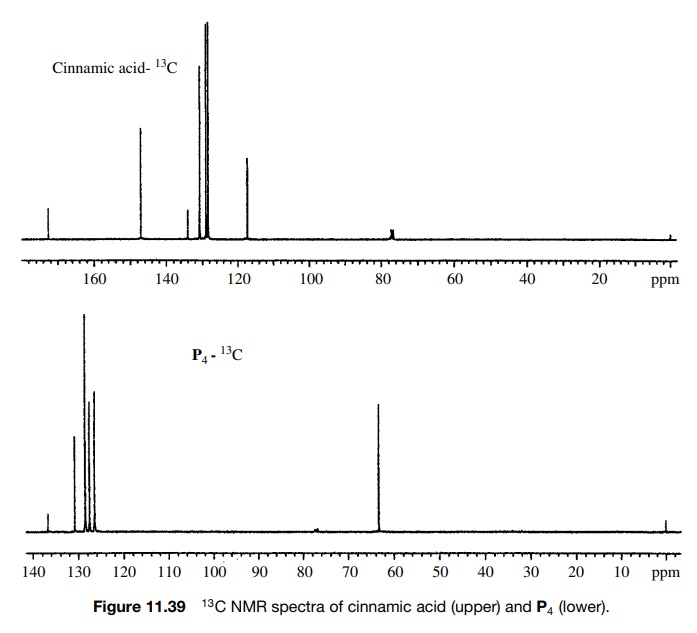
The
13C spectrum (Figure 11.39) is consistent with these structural
assign-ments as the carbonyl carbon in the reactant (172.5δ) is gone and the product contains a new signal at 63.3δ, typical for a change to sp3
hybridization and an allylic group.
In
the preceding example several types of spectroscopy are brought to bear. While
the product structure could probably be deduced from IR spectroscopy or NMR
(either 1H or 13C), the use of all three methods confirms
the assignment. It is often prudent to use more than a single technique for
structure determination so that the results reinforce each other. If a
structure assignment is not consistent with all
the data, the structure is probably incorrect.
This
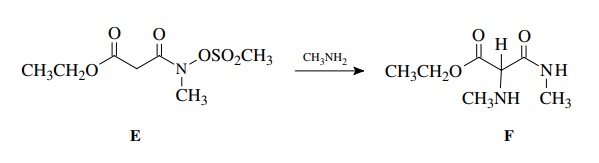
lesson is brought home in the following example taken from a recent experiment.
Ample chemical precedent suggested that the treatment of E with methyl amine should give F:
The
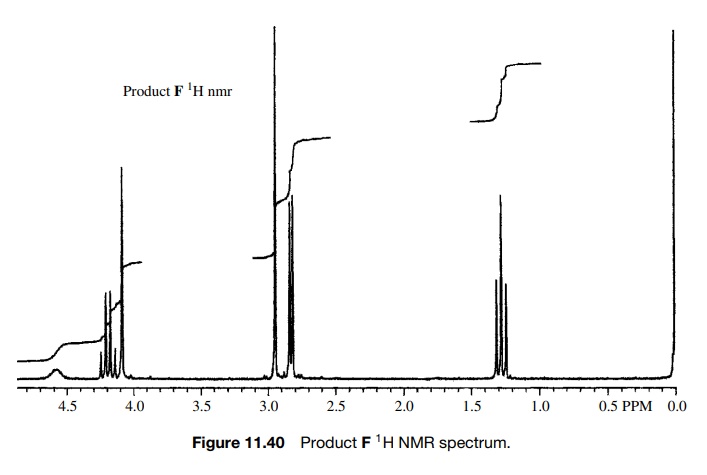
spectra of the product are shown in Figures 11.40 – 11.42. As is clear, all of
the appropriate resonances are present. In the 1H NMR (Fig. 11.40)
the amide N-methyl group is a doublet
(J = 4 Hz) at 2.76δ due to weak splitting by the amide NH proton and the amino N–CH3
group is the sharp singlet at 2.89δ
which is not split by the amine N–H proton due to rapid exchange. The C–H
methine proton is a singlet at 4.03δ
and the ethoxy group is evident by the quartet– triplet A2X3
pattern.
However,
several pieces of data just did not seem to fit. First the chemical shift of
the amino N -methyl group is at 2.90δ whereas several known compounds
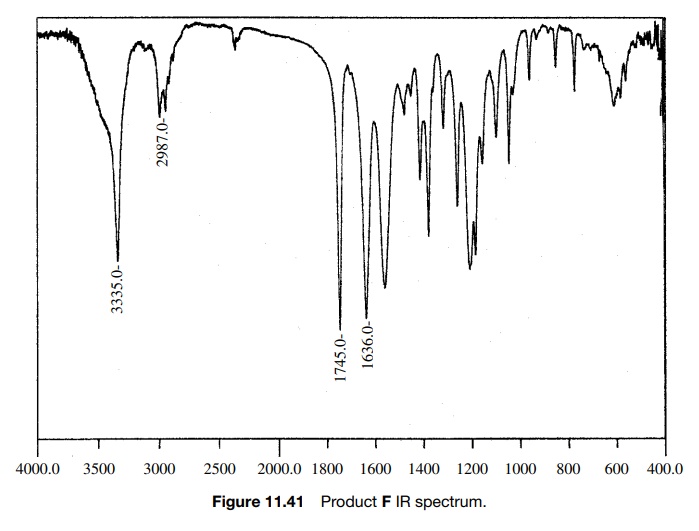
The 0.5-ppm shift might be due to the electron-withdrawing properties of the carbethoxy group, but that shift appears to be too large. In fact the chemical shift of 2.9δ is exactly that expected for an amide N -methyl, but in this case it is not split and there is already an amide N -methyl signal at 2.76δ, where it should be. Next, the integrated area of the N–H peak at 4.6δ is only 1H whereas it should account for two N–H’s which exchange. Moreover, the C–H signal at 4.03δ integrates for two protons rather than one. While one might discount the discrepancies in the integrated areas as being due to the fact that the sample was not analytically pure and while one might force fit the change in chemical shift of the amine N–CH3 group, the compound did not dissolve in acid, as was expected for the amine. Adding to the difficulty was the IR spectrum of F (Fig. 11.41). In addition to the normal ester C=O stretch at 1745 cm−1, the IR had a C=O stretch at 1636 cm−1, which is at a much lower frequency than a normal secondary amide (≈1685 cm−1).
Based on these discrepancies, the assigned structure had to be discarded. Start-ing once again from the beginning, the carbethoxy group (a) is set by the A2X3 spin system and the ester C=O stretch. The 2H singlet at 4.03δ is assigned as a two-proton CH2 group next to the ester group (b) since it has the right chem-ical shift and integrates for two protons. Another fragment which is indicated is the C(O)NHCH3 group by the chemical shift of the methyl group, its small splitting by the N–H amide proton, and the rather low C=O stretching frequency which suggests an amide group (c). What remains is an N–CH3 fragment, which can only be placed between fragments b and c. That gives the unsymmetric urea as the assigned structure of F.
This
structure fits the data in every way. The methylene group (2H) and the single
N–H proton fit the integration. Both N–CH3 groups are amide types
rather than one amino and one amide type, and both should appear at ∼2.8
– 3.0. The urea group has greater resonance stabiliza-tion, and thus the
carbonyl group has more single-bond character and comes at a lower IR frequency
than an amide. Finally F is not an amine and should not behave
chemically like one, that is, it should not dissolve in acid.
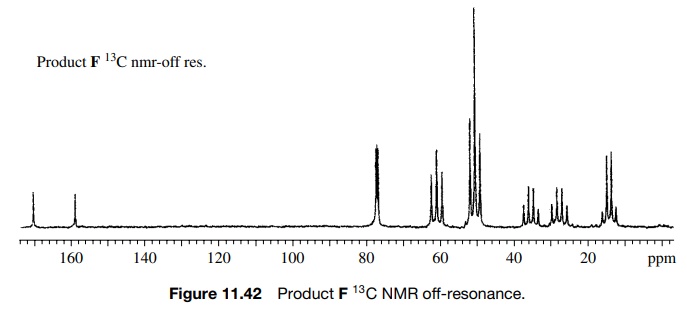
The
structure is confirmed by the off-resonance decoupled 13C spectrum
(Figure 11.42), which shows the CH2 group as a triplet 61.7δ in addition to the other expected
carbon resonances.
It
is clear that all the data must fit the structure and vice versa. If not, the
assigned structure is probably not correct. It is important to let the data
indicate the structure and not make the data fit a preconceived structure.
Often the chem-istry will suggest a structure and the data will support that
structure. However, that need not always be so, and it is necessary to always
check that the data fit the structure. New chemistry is often discovered
precisely because expected products are not supported by structural data.
Consequently new structures resulting from new chemistry are revealed.
Related Topics
